In den vorherigen Beiträgen haben wir den Bau der DNA kennengelernt und ihre Rolle bei der Zellteilung und der Herstellung von Proteinen. Die menschliche Zelle hat aber über 20.000 Gene und nicht jede Zelle unseres Körpers ist gleich. Muskelzellen brauchen andere Proteine als Nervenzellen usw. Es wäre ziemlich ineffizient, wenn alle Gene gleichzeitig in jeder Zelle exprimiert werden. Es muss also ein System vorhanden sein, dass Gene nur in den Zellen exprimiert werden, wo sie benötigt werden und auch zum richtigen Zeitpunkt. Dies wird mittels der Genregulation gesteuert. Je früher die Zelle in den Prozess der Proteinsynthese eingreift, umso weniger Energie und Aminosäuren verschwendet sie.
Die Genexpression beginnt am Promotor, wo die RNA-Polymerase bindet und dann mit der Transkription beginnt (Abb. 1). Dabei ist festzuhalten, dass in einer beliebigen Zelle zu einem beliebigen Zeitpunkt niemals alle Promotoren aktiv sind. Das deutet darauf hin, dass die Transkription von Genen selektiv erfolgen muss.
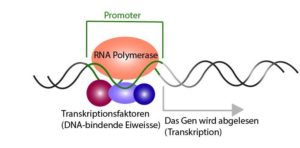
Abb. 1: RNA-Polymerase bindet an den Promotor
Bei der „Entscheidung“, welche Gene zu aktivieren sind, wirken zwei Arten von regulatorischen Proteinen mit, die an die DNA binden: Repressorproteine und Aktivatorproteine. Beide binden an den Promotor und regulieren dadurch das Gen.
Bei der negativen Regulation verhindert die Bindung des Repressorproteins an die DNA die Transkription.
Bei der positiven Regulation bindet ein Aktivatorprotein an die DNA und stimuliert dadurch die Transkription.
Genregulation bei Prokaryoten
Die ersten Erkenntnisse der Genregulation wurden bei Bakterien untersucht, vor allem bei Escherichia coli. Als normaler Bewohner des menschlichen Darms muss es sich an schnelle Veränderungen seiner chemischen Umgebung anpassen.
Der Wirt kann die Bakterien im stündlichen Wechsel mit andersartigen Nährstoffen konfrontieren (z. B. nacheinander mit Glucose und Lactose). Solche Veränderungen der Nährstoffe stellen spezifische Anforderungen an den Stoffwechsel des Bakteriums. Glucose (Traubenzucker) ist die von ihm bevorzugte Energiequelle; dieser Zucker lässt sich am einfachsten umsetzen. Fehlt Glucose als Nährstoff, kann es Lactose verdauen. Damit die Lactose von E. coli aufgenommen und verarbeitet werden kann, sind drei Enyzme erforderlich. Das Bakterium verschwendet keine Energie diese Proteine zu bilden, wenn im Wachstumsmedium keine Laktose vorhanden ist. Diese Proteine werden nur hergestellt, wenn Laktose für den Stoffwechsel verfügbar ist. Wie schafft es das Bakterium die Gene für die Lactose-abbauenden Enzyme zu aktivieren?
Dies wird mit dem sog. lac-Operon Modell erklärt (Abb. 2). Dieses besteht aus dem Promotor, als der Stelle an der die RNA-Polymerase bindet. Der zweite Bestandteil ist der Operator. Dies ist quasi gesehen der „Schalter“ für die Genaktivität. Am Operatorsequenzabschnitt findet sich eine Bindestelle für das Regulatorprotein. Die Transkription durch die RNA-Polymerase kann durch diese Blockade verhindert werden. der dritte Bestandteil sind die Strukturgene, im Falle des lac-Operons die drei Enzyme zum Abbau der Lactose. Die Strukturgene werden als lacZ, lac Y und lacA bezeichnet. Für die eigentliche Regulation verantwortlich ist das Regulator- oder Repressorprotein R. Das Gen für den Repressor (lacI) liegt außerhalb des lac-Operons. Wenn der Repressor am Operator des lac-Operons gebunden hat, wird die Transkription des Operons blockiert.
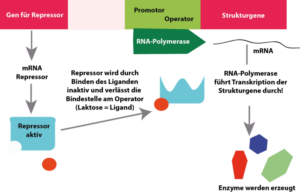
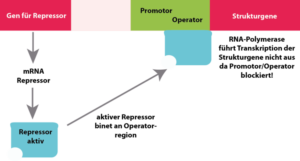
Abb. 2: Lac-Operon. Oben: Laktose bindet an das Repressorprotein und aktiviert das lac-Operon; sie Gene für die Enzyme zur Laktose-Spaltung werden aktiviert.
Unten: Bei Abwesenheit von Laktose kann das Repressorprotein an den Operator binden und blockiert das Ablesen der Gene am Lac-Operon.
Das Lac-Operon wird durch das Substrat Lactose aktiviert. Man spricht von Substratinduktion.
Das Repressorprotein enthält zwei Bindungsstellen: eine für den Operator und eine für den Induktor. Das äußere Signal aus der Umgebung, welches das lac-Operon von E. coli induziert ist Lactose. Der eigentliche Induktor ist jedoch Allolactose, ein Molekül, das aus Lactose gebildet wird, sobald Lactose in die Bakterienzelle gelangt. Ist der Induktor nicht vorhanden, bindet das Repressorprotein an den Operator. Dadurch kann die RNA-Polymerase nicht an den Promotor binden und das Operon wird nicht transkribiert. Sobald der Induktor vorhanden ist, bindet er an den Repressor und verändert dessen Konformation. Diese Änderung der dreidimensionalen Struktur verhindert, dass der Repressor an den Operator bindet. Dadurch kann die RNA-Polymerase an den Promotor binden und mit der Transkription der Strukturgene des lac-Operons beginnen. Also: Das Vorhanden sein von Lactose unterdrückt den Repressor, der nicht an den Operator binden kann und ermöglicht das Ablesen der Strukturgene. Man bezeichnet dies als Substratinduktion.
Neben der Substratinduktion gibt es auch die Endproduktrepression. Hier bindet ein Stoffwechselprodukt an ein regulatorisches Protein, das dann an den Operator binden kann und die Transkription blockiert.
Ein Beispiel dafür ist das Operon, dessen Strukturgene die Synthese der Aminosäure Tryptophan katalysieren Abb. 3). Wenn Tryptophan in der Zelle in ausreichender Konzentration vorkommt, ist es ökonomisch, die Produktion der Enzyme für die Tryptophansynthese abzuschalten. Dafür verwendet die Zelle einen Repressor, der an einen Operator im trp-Operon bindet.
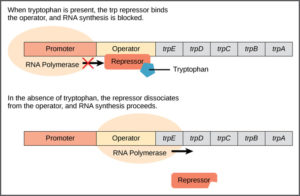
Abb. 3: trp-Operon
Aber der Repressor des trp-Operons ist normalerweise nicht an den Operator gebunden. Er bindet nur, wenn sich seine Struktur durch die Bindung mit Tryptophan verändert.
Das lac-Operon und trp-Operon sind Beispiele für die Funktion von Repressorproteine, die also an den Operator binden und die Transkription der Sturkturgene blockieren. Es gibt aber natürlich auch Aktivatorproteine, die die Transkription stimulieren.
Wie wir erfahren haben, kann der lac-Repressor nicht an den lac-Operator binden und so die Transkription zu blockieren, wenn Lactose vorhanden ist. Glucose wird jedoch von der Zelle als Energiequelle bevorzugt, sodass das lac-Operon trotz eines hohen Lactosespiegels nicht wirkungsvoll transkribiert wird, wenn zugleich der Glucosespiegel noch relativ hoch sind. Das liegt daran, dass für die effiziente Transkription des lac-Operons zusätzlich die Bindung eines Aktivatorproteins an den Operator notwendig ist.
Bei einer geringen Glucosekonzentration in der Zelle wird ein Signalweg angeschaltet, der dazu führt, dass die Konzentration von cAMP (ein sekundärer Botenstoff) zunimmt. Zyklisches AMP bindet an das Aktivatorprotein CRP (cAMP response protein). Dadurch ändert sich die Konformation von CRP, sodass das Protein an den Promotor des lac-Operons binden kann. CRP ist ein Transkriptionsaktivator, da seine Bindung dazu führt, dass die RNA-Polymerase leichter an den Promotor andockt und sich dadurch die Transkriptionsrate erhöht. In Gegenwart einer ausreichenden Menge Glucose ist die cAMP-Konzentration gering, sodass CRP nicht an den Promotor bindet, und die Transkription des lac-Operons ist weniger effizient.
Genregulation bei Eukaryoten
Damit nicht genug. Untersuchungen der Genregulation bei Bakterien haben deren Grundlagen entschlüsselt. Bei Eukaryoten und insbesondere bei mehrzelligen Lebewesen ist die Genregulation um einiges komplexer. Zum einen ist das Genom komplexer und zum anderen gibt es verschiedene Zelltypen mit verschiedenen Aufgaben und damit Genexpressionen. Bei Eukaryoten spielen die Transkriptionsfaktoren eine sehr wichtige Rolle.
Eukaryoten nutzen für die Transkription natürlich ebenfalls eine RNA-Polymerase. Es gibt jedoch in eukaryotischen Zellen hiervon drei verschiedene: Die RNA-Polymerase I, II und III. Typ I transkribiert die rRNA, Typ III die tRNA und weitere RNAs und Typ II die mRNA. Die RNA-Polymerase II kann jedoch nicht an DNA binden und selbstständig mit der Transkription beginnen. In Eukaryoten muss eine Reihe von Proteinen, sogenannte Transkriptionsfaktoren, an einen Promotor binden, bevor die RNA-Polymerase II die Transkription initiieren kann (Abb. 4).
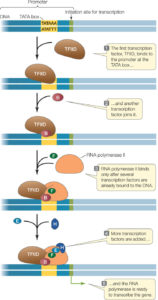
Abb. 4: Genregulation bei Eukaryoten
Der erste Transkriptionsfaktor, der bindet, wurde TFIID genannt. TFIID bindet an eine Region des Promotors, die als TATA-Box bezeichnet wird und die Sequenz TATA enthält. Die TATA-Box ist diejenige Stelle, an der die DNA anfängt zu denaturieren, sodass der Matrizenstrang zugänglich wird. Nach der TFIID-Bindung binden eine Reihe anderer Transkriptionsfaktoren und die RNA-Polymerase II einen Initiationskomplex.
Regulatorische DNA-Sequenzen wie die TATA-Box kommen in den Promotoren von zahlreichen eukaryotischen Genen vor; sie werden von Transkriptionsfaktoren erkannt, die in allen Zellen eines Organismus vorhanden sind. Andere regulatorische Sequenzen kommen nur in wenigen Genen vor und werden von spezifischen Transkriptionsfaktoren erkannt. Diese Faktoren kommen möglicherweise nur in bestimmten Zelltypen oder in bestimmten Phasen des Zellzyklus vor, oder sie werden nur als Reaktion auf Signale in der Zelle oder aus der Umgebung über Signalwege aktiviert.
Im Promotor kann sich z. B. eine DNA-Region befinden, die an Regulatorproteine binden kann. Ein Regulatorprotein bindet an die Regulatorregion und an den Transkriptionsinitiations-komplex und aktiviert ihn dadurch.
Einige regulatorische DNA-Sequenzen sind positive Elemente, die man als Enhancer bezeichnet: An sie binden Transkriptionsfaktoren, die entweder die Transkription aktivieren oder die Transkriptionsrate erhöhen. Andere regulatorische Elemente sind Silencer: An sie binden Faktoren, die die Transkription unterdrücken (Abb. 5).
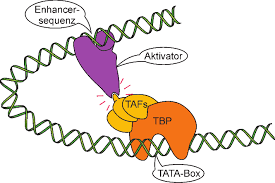
Abb. 5: Enhancer
Die meisten regulatorischen Elemente, die für die korrekte Expression eines Gens erforderlich sind, liegen innerhalb weniger Hundert Basenpaare stromaufwärts des Transkriptionsstartpunkts.
So enthält beispielsweise der Promotor des Albumingens der Maus die gesamte Information, die für die spezifische Expression des Gens in Leberzellen notwendig ist, innerhalb von 170 bp stromaufwärts des Transkriptionsstartpunkts.
Einige regulatorische Elemente können aber durchaus Tausende von Basenpaaren entfernt liegen und die Expression von mehreren nahe beieinander liegenden Genen beeinflussen. Wenn Transkriptionsfaktoren an diese Elemente binden, treten sie mit dem RNA-Polymerase-Komplex in Wechselwirkung und verursachen eine Krümmung der DNA. Die Kombination aus der Bindung von Transkriptionsfaktoren an ein Gen bestimmt die Transkriptionsrate. So enthält beispielsweise ein unreifes rotes Blutkörperchen (Erythrocyt) im Knochenmark große Mengen an ß-Globin. In diesen Zellen wirken mindestens 13 verschiedene Transkriptionsfaktoren bei der Regulation der Transkription des ß-Globin-Gens mit. Nicht alle diese Faktoren sind in anderen Zellen aktiv oder überhaupt vorhanden, beispielsweise in unreifen weißen Blutkörperchen (Leukocyten), die vom selben Knochenmark produziert werden. Das führt dazu, dass das ß-Globin-Gen in diesen Zellen nicht exprimiert wird. Es sind zwar in allen Zellen die gleichen Gene vorhanden, aber der Werdegang einer Zelle wird dadurch bestimmt, welche Gene sie wann exprimiert.
Wie aber erkennen Transkriptionsfaktoren spezifische DNA-Sequenzen? Transkriptionsfaktoren haben spezifische Domänen, die an die DNA binden können. In diesen Proteindomänen gibt es mehrere allgemeine Strukturmotive, die an DNA binden. Diese bestehen aus verschiedenen Kombinationen von Sekundärstrukturen; sie können auch besondere Komponenten wie Zink enthalten. Eines der allgemeinen Strukturmotive in DNA-bindenden Domänen ist das Helix-Turn-Helix-Motiv. Die in der großen DNA-Furche liegende „Erkennungshelix“ tritt mit den DNA-Basen sequenzspezifisch in Wechselwirkung. Die im rechten Winkel dazu nach außen zeigende Helix interagiert mit dem Zucker-Phosphat-Rückgrat, wodurch sichergestellt ist, dass die innere Helix in der richtigen Orientierung mit den Basen in Kontakt tritt. Wie kann ein Protein in der DNA eine Sequenz erkennen? Wie wir bereits wissen, bilden die komplementären Basen der DNA Wasserstoffbrücken untereinander. Sie können aber auch zusätzliche Wasserstoffbrücken mit Proteinen bilden. Auf diese Weise kann die intakte DNA-Doppelhelix von einem Proteinmotiv erkannt werden, dessen Struktur in die große oder kleine Furche hineinpasst, Aminosäuren enthält, die in das Innere der Doppelhelix hineinragen können, sowie Aminosäuren enthält, die mit den innen liegenden Basen Wasserstoffbrücken bilden können (Abb. 6).
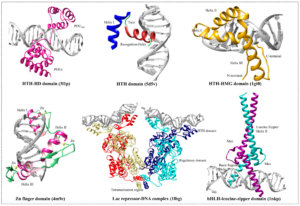
Abb. 6: Transkriptionsfaktoren bin an DNA
Epigenetik
Die Genregulation hört aber bei Transkriptionsfaktoren keineswegs auf. Immer wichtiger werden die Erkenntnisse der Epigenetik. In der Mitte des 20. Jahrhunderts formte der Entwicklungsbiologe Conrad Waddington den Begriff „Epigenetik“. Diese definierte er als „den Zweig der Biologie, der sich mit den kausalen Wechselwirkungen zwischen Genen und ihren Produkten beschäftigt, die den Phänotyp hervorbringen“. Heute definiert man die Epigenetik spezifischer und meint damit die Veränderung der Genexpression, die ohne eine Veränderung der DNA-Sequenz erfolgt. Diese Veränderungen sind reversibel, manchmal aber stabil und vererbbar. Die zwei bekanntesten Formen der Epigenetik sind DNA-Methylierung und Histon-Modifikation.
Abhängig vom Organismus werden in der DNA 1–5% der Cytosine durch Anhängen einer Methylgruppe (–CH3) zu 5-Methylcytosin modifiziert (Abb. 7). Diese kovalente Bindung wird von dem Enzym DNA-Methyltransferase katalysiert und tritt bei Säugern im Allgemeinen bei solchen Cytosinen (C) auf, die in der Sequenz stromaufwärts neben einem Guanin (G) liegen. Diese DNA-Regionen bezeichnet man als CpG-Inseln; sie kommen vor allem in Promotoren häufig vor.
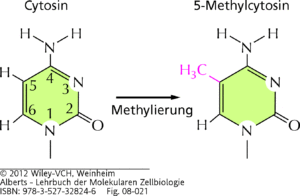
Abb. 7: Methylierung von Cytosin
Diese kovalente Veränderung der DNA ist während der Mitose erblich: Wenn die DNA repliziert wird, katalysiert eine besondere Methyltransferase die Bildung von 5-Methylcytosin im neuen DNA-Strang. Dieses Enzym führt also „Wartungsarbeiten“ durch, indem es dort, wo ein DNA-Strang Methylcytosin enthält, nach dessen Replikation die Methylierung kopiert (Abb. 8). Man spricht hier deshalb von maintenance-Enzymen („Wartungsenzymen“). Das Muster der Cytosinmethylierung kann sich auch ändern, da die Methylierung reversibel ist. Ein drittes Enzym, die Demethylase, katalysiert das Entfernen der Methylgruppe von Cytosin.
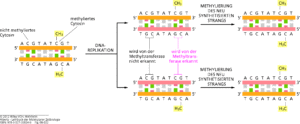
Abb. 8: Methylierung der DNA bei der Replikation
Welche Auswirkungen hat die DNA-Methylierung? Während der Replikation und der Transkription verhält sich 5-Methylcytosin wie das nicht-methylierte Cytosin. Es bildet Basenpaare mit Guanin. Aber zusätzliche Methylgruppen in einem Promotor rekrutieren weitere Proteine, die an methylierte DNA binden. Diese Proteine sind generell an der Repression der Gentranskription beteiligt. Stark methylierte Gene sind in der Tendenz inaktiver. Diese Art der genetischen Regulation ist epigenetisch, da Genexpressionsmuster ohne Veränderung der DNA-Sequenz beeinflusst werden.
Die DNA-Methylierung ist für die Entwicklung vom Ei zum Embryo von Bedeutung. Wenn beispielsweise ein Spermienzellkern eines Säugers in eine Eizelle eindringt, werden zuerst im männlichen, danach im weiblichen Genom zahlreiche Gene demethyliert. Viele Gene, die also normalerweise inaktiv sind, werden während der frühen Entwicklungsphase exprimiert.
Während sich der Embryo entwickelt und sich seine Zellen spezialisieren, werden Gene, deren Produkte für bestimmte Zelltypen nicht notwendig sind, methyliert. Diese methylierten Gene sind stillgelegt, ihre Transkription ist reprimiert. Ungewöhnliche oder anormale Ereignisse können jedoch stillgelegte Gene wieder einschalten.
Ein weiterer Mechanismus für eine epigenetische Genregulation ist die Veränderung der Chromatinstruktur (Chromatin-Remodeling). Wie wir erfahren haben, ist die DNA mit Histonproteinen zu Nucleosomen verpackt, sodass die DNA für die RNA-Polymerase und den übrigen Transkriptionsapparat unzugänglich ist. Jedes Histonprotein enthält an seinem N-Terminus einen „Schwanz“ aus etwa 20 Aminosäuren, der aus der kompakten Struktur herausragt und an bestimmten Positionen positiv geladene Aminosäuren besitzt (vor allem Lysin). Normalerweise besteht zwischen positiv geladenen Histonproteinen und der DNA, die aufgrund ihrer Phosphatgruppen negativ geladen ist, eine starke ionische Anziehungskraft.
Jedoch können Enzyme mit der Bezeichnung Acetyltransferasen an diesen positiv geladenen Aminosäuren Acetylgruppen befestigen, sodass sich die Ladung ändert (Histonacetylierung). Wenn die positive Ladung an den Histonschwänzen verringert wird, nimmt auch die Affinität der Histone für die DNA ab, sodass sich die kompakten Nucleosomen öffnen.
Weitere Chromatin-Remodeling-Proteine können an die gelockerten Histon-DNA-Komplexe binden. So wird die DNA für die Genexpression geöffnet. Histon-Acetyltransferasen können also die Transkription aktivieren (Abb. 9).
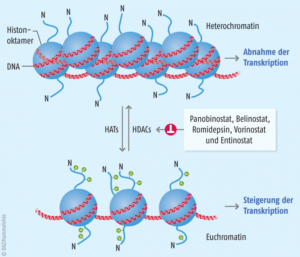
Abb. 9 Histon-Modifikation
Ein weiterer Typ von Remodelingproteinen sind die Histon-Deacetylasen. Sie können Acetylgruppen von Histonen entfernen und reprimieren so die Transkription.
Acetylierung ist nicht die einzige Form der Modifikation von Histonen, durch den die Genaktivierung und die Genrepression beeinflusst werden. So hängt beispielsweise die Histonmethylierung (die Sie nicht mit der Methylierung der DNA verwechseln sollten) mit der Inaktivierung von Genen zusammen. Auch die Histonphosphorylierung beeinflusst die Genexpression.
Die spezifische Wirkung hängt davon ab, welche Aminosäure modifiziert wird. Alle diese Effekte sind reversibel, sodass die Aktivität eines eukaryotischen Gens durch sehr komplexe Muster der Histonmodifikation bestimmt sein kann.
Epigenetische Veränderungen werden z. B. durch Umweltfaktoren beeinflusst. Z. B. besteht ein Bienenstaat aus verschiedenen Kasten: unfruchtbare Arbeiterbienen, fruchtbare Königinnen, etc. Was bestimmt aber, ob aus einer Bienenlarve eine Königin oder Arbeiterin wird? Sicherlich nicht die Erbfolge der DNA, denn weibliche Honigbienen besitzen alle eine sehr ähnliche genetische Ausstattung. Während des Larvenstadiums füttern die Arbeiterinnen jedoch bestimmte Weibchen im Stock mit Gelée royale (eine proteinreiche Substanz), wodurch sich bei zahlreichen Genen die Expression ändert. Die jungen Königinnen werden viel größer als die Arbeiterinnen und wird geschlechtsreif (Abb. 10).
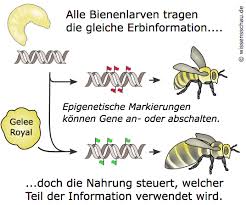
Abb. 10: Epigenetik der Bienen
Epigenetische Veränderungen sind zwar reversibel, aber viele dieser Veränderungen, etwa DNA-Methylierung und Histonacetylierung, können die Genexpressionsmuster in einer Zelle dauerhaft verändern.
Lange Zeit ging man davon aus, dass diese epigenetischen Veränderungen nicht auf die Keimzellen übertragen werden, denn ein Großteil der epigenetischen Marker werden bei der Bildung von Keimzellen während der Meiose gelöscht. Mittlerweile ist aber bekannt, dass einige epigenetische Marker auch in den Keimzellen vorhanden bleiben oder neu entstehen und zumindest auch über einige Generationen stabil bleiben. Dieses Feld der Vererbung erworbener Eigenschaften ist ziemlich spannend und liefert sicherlich Material für genügend für weitere Beiträge. Doch die Genregulation hat immer noch kein Ende, zwei weitere Prozesse sollen noch vorgestellt werden, die nach der Transkription stattfinden: alternatives Spleißen und die RNA-Interferenz.
Alternatives Spleißen und RNA-Interferenz
Im Beitrag zur Proteinbiosynthese haben wir gelernt, dass Gene in Introns und Exons geteilt sind. Nach der Transkription werden die Introns aus der prä-mRNA ausgeschnitten und die Exons, die die Informationen für die Aminosäurenabfolge haben, zusammengefügt. Eine Variation dieses Vorgangs wird als alternatives Spleißen bezeichnet (Abb. 11). Das funktioniert nach folgendem Prinzip:
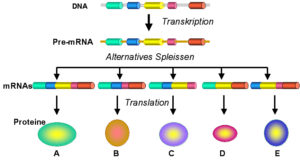
Abb. 11: Alternatives Speißen
Nehmen wir mal an ein Gen besteht aus 5 Exons und 4 dazwischenliegenden Introns. Beim Spleißen würden die 4 Introns ausgeschnitten werden, sodass die 5 Exons übrigbleiben. Beim normalen Spleißen würden die Exons in der „richtigen“ Reihenfolge 1-5 hintereinander zusammengefügt werden. Beim alternativen Spleißen können die Exons jedoch unterschiedlich kombiniert werden. so wäre eine Reihenfolge von Exon 5,1,4,2,3 möglich. Es wäre aber auch denkbar, dass nicht alle Exons Verwendung finden würden, z. B. würde für ein Genprodukt nur die Exone 1,2 und 3 benötigen. Dieses alternative Spleißen ermöglicht, dass man zwar nur 20.000 Gene hat, aber dennoch wesentlich mehr Proteine bilden kann. Hierbei handelt es sich aber um einen zielgerichteten Vorgang, das sowohl durch regulatorische Elemente in der RNA-Sequenz, an die spezifische Proteine binden, als auch durch Sekundärstrukturen reguliert wird, die sich mittels Hybridisierung von Nucleotiden im einzelsträngigen RNA-Molekül bilden. Wie aktuelle Untersuchungen zeigen, unterliegt etwa die Hälfte aller menschlichen Gene einem alternativen Spleißen. Mithilfe des alternativen Spleißens lassen sich möglicherweise auch unterschiedliche Komplexitätsniveaus von Lebewesen erklären.
Obwohl beispielsweise die Genome von Mensch und Schimpanse etwa gleich groß sind, kommt es im menschlichen Gehirn zu mehr alternativen Spleißvorgängen als beim Schimpansen.
Wir haben verschiedene Typen der RNA kennengelernt: mRNA, tRNA und rRNA. Es gibt jedoch eine Reihe weiterer RNA-Moleküle, die teilweise von den nicht-proteincodierenden DNA-Sequenzen exprimiert werden. Die RNA-Produkte dieser Regionen sind häufig sehr klein und deshalb schwer nachzuweisen. Man bezeichnet die winzigen RNA-Moleküle bei Prokaryoten und Eukaryoten als mikroRNA (miRNA, Abb. 12).
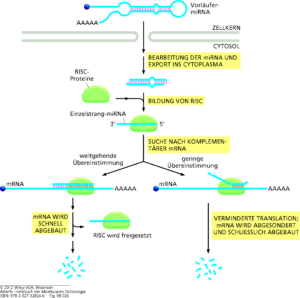
Abb. 12: miRNA. Die miRNA-Vorstufe wird weiter bearbeitet und es entsteht eine reife miRNA. Diese lagert sich mit einer Gruppen von Proteinen zu einem Komplex zusammen, den man RISC nennt. Die miRNA leitet den RISC dann zu mRNAs mit einer komplementären Nukleotidsequenz. Je nachdem, wie groß der komplementäre Bereich ist, wird die Ziel-mRNA entweder schnell von einer Nuklease des RISC abgebaut oder an einen Ort im Cytoplasma transportiert, an dem andere zelluläre Nukleasen sie zerstören.
Die internationale miRNA-Datenbank „miRBase“ hatte im Frühjahr 2018 rund 30.000 Einträge, davon bezogen sich etwa 2660 auf das menschliche Genom. Diese miRNAs sind etwa 22 Basen lang und es gibt für jede mehrere Ziel-mRNAs. miRNAs werden in Form längerer Vorstufen transkribiert, die sich zu doppelsträngigen mRNA-Molekülen falten und dann durch eine Reihe von Reaktionsschritten zu einzelsträngigen miRNAs prozessiert werden. Ein Proteinkomplex lenkt die miRNA zu ihrer Ziel-mRNA, wo die Translation blockiert wird. Diese bemerkenswerte Konservierung des miRNA-vermittelten Gen-Silencings deutet darauf hin, dass es sich um einen evolutionär sehr alten Mechanismus von großer biologischer Bedeutung handelt.
Neben den miRNAs gibt es eine weitere Art von RNA-Molekülen, die auf ähnliche Weise wirken: kleine interferierende RNAs (small interfering RNAs, siRNAs, Abb. 13). Diese treten häufig bei Virusinfektionen auf, wenn zwei komplementäre Stränge eines Virusgenoms transkribiert werden. Dabei entstehen große doppelsträngige RNAs, die dann wie im Fall der miRNAs in kürzere, einzelsträngige Sequenzen umgewandelt werden.
Abb. 13: siRNA. siRNAs zerstören fremde RNAs. Doppelsträngige RNAs von einem Virus oder einem transponierbaren genetischen Element werden zuerst von einer Nuklease gespalten, die Dicer genannt wird. Die so entstandenen doppelsträngigen Fragmente werden in RISCs aufgenommen. Im RISC wird ein Strang der Doppelhelix verworfen und der andere Strang wird verwendet, um komplementäre RNAs aufzuspüren und zu zerstören. Dieser Mechanismus bildet die Grundlage für die RNA-Interferenz (RNAi).
Diese binden an die Ziel-RNA und verursachen deren Abbau. Kleine interferierende RNAs leiten sich auch aus Transposonsequenzen ab, die in den Genomen der Eukaryoten weit verbreitet sind. Demnach hat sich wahrscheinlich das Abschalten von Genen durch siRNAs in der Evolution als Abwehrmechanismus gegen die Translation von Virus- und Transposonsequenzen entwickelt. miRNAs und siRNAs sind ähnliche Moleküle, die von den gleichen zellulären Enzymen prozessiert werden. Es gibt jedoch einen wichtigen Unterschied:
miRNAs werden von DNA-Sequenzen synthetisiert, die von den Zielsequenzen getrennt sind, während siRNAs gegen die Sequenz gerichtet sind, aus der sie stammen.
Die Bildung einer mRNA führt übrigens nicht automatisch zur Translation. Oft wird wesentlich mehr RNA gebildet als Proteine tatsächlich vorhanden sind. Auch der umgekehrte Fall kann eintreten: es gibt mehr Proteine als mRNA. Es gibt also Prozesse in der Zelle, die die Translation regulieren, sowie die Verweildauer eines Proteins in der Zelle.
Es gibt eine Reihe verschiedener Mechanismen, durch welche die Translation der mRNA reguliert werden kann. Zum einen kann die Translation durch siRNAs und miRNAs blockiert werden, zum anderen kann die Cap-Struktur der RNA beeinflusst werden. Dies ist ein chemisch modifiziertes Guanosintriphosphat (GTP). Dadurch wird später bei der Translation die Bindung der mRNA an das Ribosom unterstützt und die mRNA ist vor einem Abbau geschützt. Eine mRNA, die ein nichtmodifiziertes GTP-Molekül als Cap-Struktur enthält, wird nicht translatiert. In einem anderen System blockieren Repressorproteine die Translation direkt. So bindet beispielsweise das Protein Ferritin in Säugerzellen freie Eisen(II)-Ionen (Fe2+). Wenn Eisen im Überschuss vorhanden ist, nimmt die Ferritinsynthese erheblich zu, aber die Menge der Ferritin-mRNA bleibt konstant (Abb. 14).
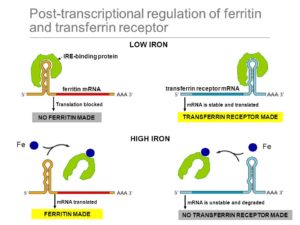
Abb. 14: Regulation der Ferritin-Synthese
Offenbar ist die Zunahme der Ferritinsynthese auf eine erhöhte Translationsrate der mRNA zurückzuführen. Wenn der Eisenspiegel in einer Zelle gering ist, bindet ein Repressorprotein der Translation an die nichtcodierende 5′-Region der Ferritin-mRNA und hemmt die Translation, indem es die Bindung an das Ribosom verhindert. Wenn der Eisenspiegel steigt, bindet ein Teil der überschüssigen Fe2+-Ionen an den Repressor, dessen dreidimensionale Struktur sich daraufhin ändert, sodass er sich von der mRNA ablöst und die Translation voranschreiten kann.
Wir wissen nun über einige Mechanismen der Genregulation Bescheid – es gibt sicherlich noch andere. Es konnte gezeigt werden, dass die Genexpression von äußeren Faktoren abhängt. Aber wie kommen die äußeren Reize zum einen an die Zelle und zum anderen an die entsprechenden Gene? Also wie passiert es z. B., dass durch das Füttern mit Gele royale die Genexpression bei Bienenlarven sich ändert? Dies wird mittels Zellkommunikation geklärt.