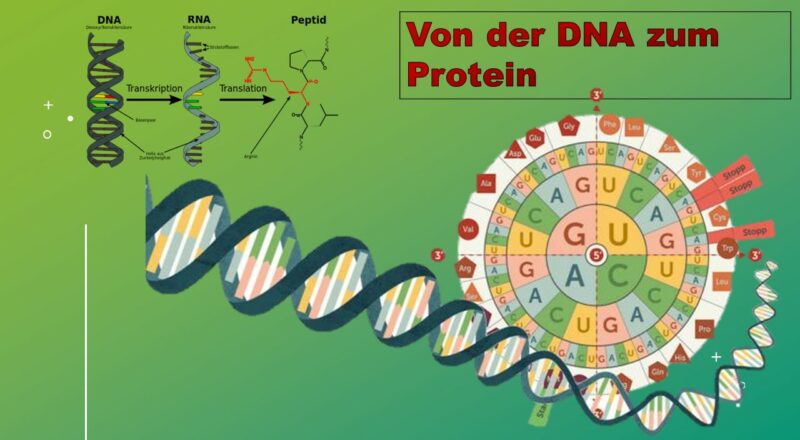
In unserem Kapitel über den Aufbau der DNA stellten wir fest, dass diese Informationen für den Aufbau von Proteinen enthält, die Gene. Proteine haben wir ebenfalls kennengelernt. Proteine können quasi gesehen als Genprodukte verstanden werden. Werden Gene abgelesen und in Proteine übersetzt spricht man auch von Genexpression. Das zugehörige Verb lautet exprimieren (Abb. 1). Die Gesamtheit der Gene wird als Genotyp bezeichnet, die Merkmale für die sie codieren – als Phänotyp.
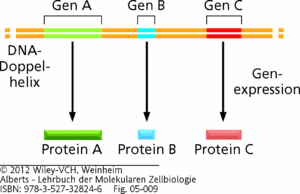
Abb. 1: Genexpression
Ein-Gen-Ein-Enzym-(Polypeptid)-Hypothese
Schon in den 1940er Jahren führten George Beadle und Edward Tatum Untersuchungen durch, um die Phänotypen von Neurospora chemisch zu bestimmen (Abb. 2). Neurospora gehört zu den Pilzen, die die meiste Zeit ihres Lebens haploid sind. Beadle und Tatum stellten die Hypothese auf, dass die Expression eines spezifischen Gens zur Aktivität eines spezifischen Enzyms führt. Nun wollten sie diese Hypothese direkt testen. Sie ließen Neurospora auf einem Nährmedium wachsen, das Saccharose, Mineralsalze und Biotin enthielt, das einzige Vitamin, das die Wildtypform von Neurospora nicht selbst produzieren kann. Auf diesem Minimalmedium konnten die Enzyme des Neurospora-Wildtyps alle Stoffwechselreaktionen katalysieren, die für das Wachstum erforderlich sind.
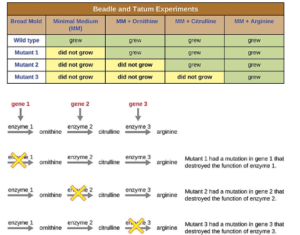
Abb. 2: Experiment von Beadle & Tatum
Die Wissenschaftler behandelten die Wildtypform von Neurospora mit Röntgenstrahlen, die als Mutagen wirken. Ein Mutagen ist ein Faktor, der DNA schädigt und Mutationen hervorruft. Nach der Behandlung mit Röntgenstrahlen konnten einige Neurospora-Stämme nicht mehr auf Minimalmedium wachsen. Diese mutierten Stämme wuchsen nur noch, wenn man dem Medium spezifische Nährstoffe zusetzte, etwa bestimmte Vitamine.
Beadle und Tatum stellten die Hypothese auf, dass diese genetischen Stämme in den Genen, die für die Produktion von Enzymen verantwortlich sind Mutationen trugen. Jede Mutation führte in einem bestimmten Gen zur Funktionslosigkeit des Enzyms. Dies ist die sogenannte Ein-Gen-ein-Enzym-Hypothese, bzw. erweitert als Ein-Gen-ein-Protein-Hypothese (Abb. 2).
Die Ein-Gen-ein-Protein-Hypothese hat mit zunehmenden Erkenntnissen der molekularen Biologie mehrere Erweiterungen erfahren. Viele Proteine, darunter auch zahlreiche Enzyme, bestehen aus mehr als einer Polypeptidkette oder Untereinheit, das heißt, sie besitzen eine Quartärstruktur. Es ist also korrekter, von der Beziehung Ein-Gen-ein-Polypeptid zu sprechen.
Die Funktion eines Gens besteht darin, die Information für die Produktion eines einzigen spezifischen Polypeptids zu liefern.
Genexpression und zentrales Dogma der Molekularbiologie
Doch die Gene werden nicht direkt in Protein übersetzt, sondern über einen Zwischenweg, die Bildung von RNA. Die RNA haben wir ebenfalls kennengelernt. Sie ist eine Nukleinsäure wie die DNA, hat aber als Zucker die Ribose und statt der Base Thymin die Base Uracil.
Werden die Informationen aus der DNA in Proteine „übersetzt“, spricht man von der Eiweißsynthese oder Proteinbiosynthese. Diese Verläuft in zwei Teilprozessen, Transkription und Translation, die wir hier kennenlernen werden.
Aus dieser Erkenntnis lässt sich das sog. „zentrale Dogma“ der Molekularbiologie schließen (Abb. 3):
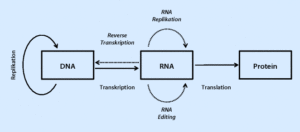
Abb. 3: Zentrales Dogma der Molekularbiologie
Der Informationsfluss fließt von der DNA zur RNA zum Protein. Der Begriff „zentrales Dogma“ ist unglücklich gewählt, wirkt es doch fast nach Glaubenssätzen der katholischen Kirche. Aber im Prinzip trifft diese Informationsrichtung in der Proteinbiosynthese zu und wir werden sehen, dass sich aus der Struktur des Proteins nicht die DNA-Sequenz genau ableiten lässt. Aber mittlerweile hat dieses „zentrale Dogma“ einige Revidierungen erfahren. So ist bei manchen Viren bekannt, dass die RNA in DNA umwandeln können, wenn sie das Enzym Reverse Transkriptase besitzen. Das trifft z. B. auf Retroviren wie das HIV zu. RNA-Viren können auch ohne DNA Proteine bilden, was wir von Corona-Viren wissen. Und mittlerweile ist auch bekannt, dass einige Proteine die Information zur Bildung von Proteinen haben. Das ist bei Prionen bekannt. Prionen, die eine spezielle Faltung im Vergleich zum ursprünglichen Normalprotein haben, sind in der Lage andere Proteine in Prionen umzuwandeln. Rinderwahn und die Creutzfeldt-Jakob-Krankheit werden von Prionen verursacht. Proteine haben zwar nicht die Information DNA zu bilden, können aber mittels Genregulation Einfluss auf die Genexpression nehmen. Die Genregulation wird ein anderes Mal behandelt.
Transkription
Damit ein Protein-kodierendes Gen exprimiert werden kann, muss es zuerst transkribiert werden. Bei der Transkription wird der Code in der DNA des Gens in einen komplementären Code in einem RNA-Molekül umgewandelt. Diese RNA wird als Boten-RNA, Messenger-RNA oder kurz mRNA bezeichnet. Die Transkription erfolgt in drei Schritten: Initiation, Elongation und Termination (Abb. 4).
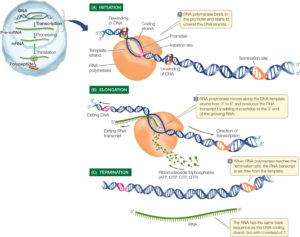
Abb. 4: Transkription
Initiation
Auch für die Transkription braucht es Enzyme. Hier ist das Enzym RNA-Polymerase entscheidend. Die Initiation der Transkription erfolgt an einem Promotor, einer Region auf der DNA, an der die RNA-Polymerase bindet. Die RNA-Polymerase bindet an den Promotor und beginnt, die DNA abzuwickeln. Nun liegt der DNA-Doppelstrang offen und die Information für die Synthese liegt frei. Aber welcher der beiden DNA-Stränge wird abgelesen, dient also als Matrize für die RNA-Synthese? Wie die DNA-Polymerasen nutzen auch die RNA-Polymerasen den 3‘-5‘-Strang der DNA als Matrize zur Synthese von mRNA, welche dann in 5‘-3‘-Richtung gebildet wird. Diesen Strang bezeichnet man auch als Anti-Sense-Strang oder codogener Strang. Der andere DNA-Strang (5‘-3‘) ist der Sense-Strang oder codierender Strang und hat die Basensequenz des entsprechenden Gens.
Mit dem Anti-Sense-Strang als Matrize für die RNA-Synthese entsteht eine RNA, deren Nucleotidsequenz identisch ist mit der des DNA-Sense-Strangs. Die RNA wird also komplementär zu ihrer DNA-Matrize synthetisiert und hat dadurch die gleiche Sequenz wie der Sense-Strang, also um das eigentliche Gen. Damit ist die betreffende DNA-Information direkt in der RNA gespeichert (Abb. 5).
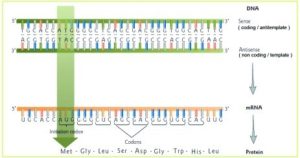
Abb. 5: Sense – und Anti-Sense-Strang der DNA
Im Gegensatz zu DNA-Polymerasen benötigen RNA-Polymerasen keinen Primer.
Teil eines jeden Promotors ist die Initiationsstelle, an der die Transkription beginnt. Nucleotidgruppen, die „stromaufwärts“ der Initiationsstelle liegen, unterstützen die RNA-Polymerase bei der Bindung.
Weitere Proteine, die an spezifische DNA-Sequenzen und an die RNA-Polymerase binden können, tragen dazu bei, die RNA-Polymerase zum Promotor zu dirigieren. Diese Proteine, die man bei den Prokaryoten als Sigmafaktoren und bei den Eukaryoten als Transkriptionsfaktoren bezeichnet, haben einen Einfluss darauf, welche spezifischen Gene in einer Zelle zu einem bestimmten Zeitpunkt exprimiert werden.
Jedes Gen besitzt zwar einen Promotor, aber nicht alle Promotoren sind identisch. Einige Promotoren sind bei der Initiation der Transkription effizienter als andere. Transkriptionsfaktoren und Promotoren-Effizienz werden wir bei der Genregulation näher kennenlernen.
Elongation:
Sobald die RNA-Polymerase an den Promotor gebunden hat, beginnt sie mit der Elongation. Die DNA wird bei jedem Schritt über einen Abschnitt von etwa zehn Basenpaaren entwunden, und die RNA-Polymerase liest den Anti-Sense-Strang (er verläuft in 3‘-5‘-Richtung) ab. Die neue RNA wird beginnend mit der ersten Base, die das 5‘-Ende bildet, durch Anfügen von Nucleotiden, die zur DNA-Sequenz komplementär sind, zum 3‘-Ende hin verlängert. Dieser Prozess kostet Energie. Bei den Einzel-Nukleotiden für die mRNA, die die RNA-Polymerase nutzt, handelt es sich um Ribonucleosidtriphosphate: Durch die Abspaltung von zwei Photsphatgruppen wird Energie frei und die nun mit nur einem Phosphat bestückten Einzel-Nukleotide bilden die mRNA.
Termination:
Genauso wie Initiationsstellen im DNA-Matrizenstrang den Startpunkt der Transkription festlegen, bestimmen auch bestimmte Basensequenzen ihre Termination. Es gibt zwei Mechanismen für die Beendigung der Transkription. Bei manchen Genen faltet sich das neu entstandene Transkript auf sich selbst zurück und bildet interne Wasserstoffbrücken zwischen den Basen. So entsteht eine Schleife, durch die das neu gebildete Transkript von der DNA-Matrize und der RNA-Polymerase abfällt. In anderen Fällen bindet ein Protein an spezifische Sequenzen auf dem Transkript und bewirkt so, dass sich das Transkript von der DNA-Matrize löst.
Genetischer Code
Der genetische Code ist der Schlüssel, mit dessen Hilfe die Nucleotidsequenz einer mRNA, die einem Gen entspricht, in eine Aminosäuresequenz, aus der das von dem Gen codierte Protein besteht, übersetzt (translatiert) wird (Abb. 6). Das heißt, der genetische Code legt fest, welche Aminosäuren nacheinander verwendet werden, um eine bestimmte Polypeptidkette zu bilden.
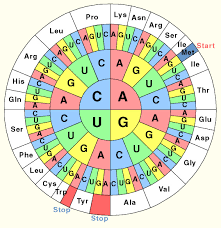
Abb. 6: genetischer Code
Man kann sich den genetischen Code als eine spezielle Folge nicht-überlappender Buchstabenkombinationen vorstellen. Dabei werden die einzelnen Basen der RNA: Adenin, Cytosin, Uracil und Guanin durch die Buchstaben A, C, U und G abgekürzt. Der genetische Code der RNA ist komplementär zum Anti-Sense-Strang der DNA, der als Matrize diente. Damit ist der Code der RNA identisch zur Basensequenz des Sense-Stranges, der die Information für das Gen (die Aminosäurenabfolge) hat. Drei einzelne Nukleotide hintereinander bilden einen Code, quasi die Information für eine Aminosäure. So steht z. B. die Kombination der Buchstaben AAA (3x Adenin) für die Aminosäure Lysin. Der genetische Code ist in den 1960er Jahren entschlüsselt worden. Die damalige Fragestellung war, Wie lassen sich mit einem Alphabet, das nur aus vier Buchstaben besteht, mindestens 20 Codewörter schreiben? Oder anders ausgedrückt, wie können die vier RNA-Basen (A, U, G und C) 20 verschiedene Aminosäuren codieren? Da es nur vier Buchstaben gibt, hätte ein Ein-Buchstaben-Code nur vier Aminosäuren codieren können, ein Zwei-Buchstaben-Code nur 4 * 4 = 16. Aber ein Triplettcode mit 4*4*4 = 64 Codons würde mehr als genügen, um 20 Aminosäuren zu codieren.
Nun haben wir es aber so, dass wir 64 Codes haben, aber nur 20 Aminosäuren brauchen. Das führt dazu, dass manche Aminosäuren von mehr als nur einem Code bestehen. Der genetische Code ist somit redundant (manche sagen auch „degeneriert“). Die erwähnte Aminosäure Lysin wird z. B. nicht nur durch die Kombination aus AAA codiert, sondern auch die Kombination AAG. Leucin hat sogar 6 Codes.
Daraus ist übrigens ersichtlich, weshalb eine Rückführung der Aminosäurensequenz zur DNA-Sequenz nicht so einfach ist, da mehrere Aminosäuren für mehrere Basensequenzen stehen können.
Von den 64 Codes stehen 61 für Aminosäuren zur Verfügung. Wobei die Aminosäure Methionin durch den Code AUG codiert wird und als Startcodon für die Initiation der Transkription dient. D. h. jede Proteinsynthese startet mit Methionin als Erkennungssequenz für das Ablesen der Gene. Die drei weiteren Codes (UAA, UAG und UGA) sind sog. Stopcodons (Terminationssignale).
Fast alle Spezies auf diesem Planeten verwenden denselben genetischen Code. Dieser Code wurde in der Evolution des Lebens nahezu durchweg vollständig beibehalten und muss daher immens alt sein. Einige Ausnahmen sind jedoch bekannt: So weicht beispielsweise der Code der mitochondrialen DNA und der Chloroplasten-DNA geringfügig von dem Code der Prokaryoten und der Zellkern-DNA der eukaryotischen Zellen ab. UAA und UAG sind dort keine Stoppcodons, sondern codieren Glutamin. Die Bedeutung dieser Unterschiede ist noch unbekannt. Auf jeden Fall ist die Zahl der bekannten Ausnahmen sehr gering.
Marshall Nirenberg und Heinrich Matthaei schafften 1961 den ersten Durchbruch zur Entschlüsselung des genetischen Codes, als sie anstelle der komplexen natürlichen mRNA ein einfaches, synthetisches Polynucleotid als Informationsträger verwenden konnten. Sie begannen mRNA-Moleküle zu synthetisieren, die nur aus einer einzigen Art von Nucleotiden bestanden. So besteht eine Poly(U)-mRNA nur aus Uracilnucleotiden.
Nirenberg und Matthaei wollten nun durch eine Translation im Testgefäß das Polypeptid ermitteln, das diese künstliche mRNA codierte. Ihr Experiment führte zur Identifizierung der ersten Codons. Andere Wissenschaftler fanden bald darauf auch den übrigen Code heraus.
Prozessierung & Splicing
Da alle Lebewesen den genetischen Code gemeinsam haben, könnte man jetzt annehmen, dass der Vorgang der Genexpression bei Eukaryoten und Prokaryoten sehr stark übereinstimmt.
Für den grundlegenden Mechanismus trifft das auch zu, in den Details gibt es jedoch Unterschiede, die wir hier kennenlernen werden.
Die Sequenz einer mRNA, die in einer Zelle an einem Ribosom in ein Polypeptid übersetzt wird, entspricht sowohl bei Prokaryoten als auch bei Eukaryoten einer bestimmten Gensequenz in der DNA dieses Lebewesens. Bei den Eukaryoten befinden sich aber Abschnitte innerhalb eines Gens, welche nicht für die Proteinsynthese verwendet werden.
Gene bei Eukaryoten liegen oft nicht als „ununterbrochene“ Kette vor, sondern sind „zerstückelt“ in „Introns“ und „Exons“. Exons werden die codierten Elemente genannt, die also die Information für den Aufbau der Proteine haben, die Introns sind die nicht codierenden Bereiche (Abb. 7).
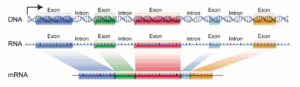
Abb. 7: Introns & Exons
Die nichtcodierenden Introns werden zwar transkribiert – es entsteht erst mal eine prä-mRNA mit den transkribierten Introns und Exons, aber dann im Zellkern aus der Prä-mRNA entfernt. Nur die Exons verbleiben so schließlich in der bearbeiteten mRNA, die den Zellkern verlässt. Den Schritt, in dem die Introns entfernt werden, bezeichnet man als RNA-Spleißen. Das RNA-Spleißen erfolgt durch das Spleißosom. Dieser RNA-Protein-Komplex entfernt die Introns und verknüpft die Exons (Abb. 8).
Sobald eine Prä-mRNA transkribiert wird, wird sie schnell von mehreren Komplexen gebunden, die als Kleine Kern-Ribonukleoprotein-Partikel (small nuclear ribonucleoprotein particles) oder kurz snRNPs (ausgesprochen „Snurps“) bezeichnet werden. Wie der Name schon sagt, enthalten snRNPs RNAs und Proteine. Die snRNPs sind für das Spleißen von Introns aus Prä-mRNAs verantwortlich.
snRNPs binden an Stellen in einer Prä-mRNA an oder nahe den Intron-Exon-Grenzen. Diese als Konsensussequenzen bezeichneten Stellen enthalten Nukleotidsequenzen, die von den meisten Prä-mRNAs gemeinsam genutzt werden. Die snRNPs enthalten RNA-Moleküle, die durch komplementäre Basenpaarung an diese Konsensussequenzen binden können.
Die snRNPs bilden schließlich einen großen Komplex, der als Spleißosom bezeichnet wird. Während sich das Spleißosom bildet, tritt das Intron aus. Das Spleißosom schneidet die Prä-mRNA an einer Intron-Exon-Grenze, wo es eine reaktive freie Hydroxylgruppe (-OH) am Exon hinterlässt. Das Spleißosom verwendet diese Hydroxylgruppe, um das andere Ende des Introns anzugreifen, und entfernt dabei das Intron und verbindet die Exons miteinander, wobei ein reifes mRNA-Molekül gebildet wird.
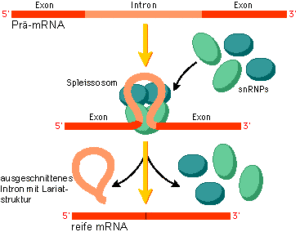
Abb. 8: Splicing & Spleißosom
Neben dem Spießen gibt es noch weitere Veränderungen der mRNA. Denn diese muss ja ungehindert den Zellkern verlassen und an die Ribosomen im Zellplasma andocken, ohne abgebaut zu werden (Abb. 9).
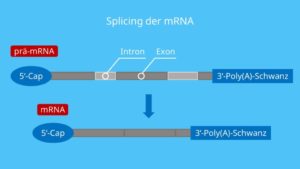
Abb. 9: Prozessierung der mRNA
Am 5‘-Ende der Prä-mRNA wird während der Transkription eine 5‘-Cap-Gruppe angehängt. Dies ist ein chemisch modifiziertes Guanosintriphosphat (GTP). Dadurch wird später bei der Translation die Bindung der mRNA an das Ribosom unterstützt und die mRNA ist vor einem Abbau geschützt.
Am 3‘-Ende der Prä-mRNA wird ein Poly(A)-Schwanz angehängt. Die Transkription eines Gens endet stromabwärts des Terminationscodons in der DNA. Bei den Eukaryoten liegt normalerweise hinter dem letzten Codon in der Nähe des 3‘-Endes der Prä-mRNA eine sogenannte Polyadenylierungs-Sequenz (AAUAAA). Diese Sequenz wirkt als Signal auf ein Enzym, das die Prä-mRNA abschneidet. Unmittelbar nach diesem Schnitt hängt ein anderes Enzym 100–300 Adenin-Nucleotide an das 3‘-Ende der Prä-mRNA. Dieser Poly(A)-Schwanz unterstützt den Export der mRNA aus dem Zellkern und ist für die Stabilität der mRNA von Bedeutung.
Translation
Die Transkription und posttranskriptionelle Vorgänge bringen eine mRNA hervor, die in eine Aminosäuresequenz translatiert werden kann. Die Information der mRNA wird in eine Aminosäurensequenz übersetzt. In der Biologie ist dieser Übersetzer eine besondere Art von RNA-Molekül, das man als Transfer-RNA (tRNA) bezeichnet. Um eine genaue Übersetzung sicherzustellen – das heißt, das Protein wird genau nach den Vorgaben der mRNA synthetisiert – müssen die tRNAs erstens jedes Codon korrekt ablesen und zweitens die zugehörigen Aminosäuren zum Codon bringen. Die Translation findet im Cytoplasma an den Ribosomen statt, sie werden häufig als Proteinfabriken bezeichnet.
Für jede der 20 Aminosäuren gibt es mindestens ein spezifisches tRNA-Molekül. Jede tRNA besitzt drei Funktionen, die durch ihre Struktur und ihre Basensequenz bestimmt werden (Abb. 10).
![]()
Abb. 10: tRNA
- tRNAs binden spezifische Aminosäuren. Jede tRNA bindet an ein Enzym, das spezifisch eine der 20 Aminosäuren an sie koppelt. Die kovalente Bindung mit der Aminosäure erfolgt am 3‘-Ende der tRNA. Wenn die tRNA eine Aminosäure trägt, bezeichnet man sie als „beladen“. Die dafür zuständigen Enzyme werden als Aminoacyl-tRNA-Synthetasen bezeichnet. Jedes dieser Enzyme ist für eine Aminosäure und die zugehörige tRNA spezifisch. Bei der Reaktion wird ATP verbraucht, sodass zwischen einer tRNA und der Aminosäure eine sehr energiereiche Bindung entsteht. Die Energie dieser Bindung dient später dazu, die Peptidbindung zwischen der Aminosäure und der wachsenden Polypeptidkette zu bilden.
- tRNAs binden an mRNA. Etwa in der Mitte der Polynucleotidkette der tRNA befindet sich ein Triplett, das man als Anticodon bezeichnet. Es ist komplementär zum mRNA-Codon für die zugehörige Aminosäure, die die betreffende tRNA trägt. Wie die beiden Stränge der DNA binden auch das Codon und das Anticodon über Wasserstoffbrücken aneinander.
- tRNAs treten mit dem Ribosom in Wechselwirkung. Das Ribosom enthält an seiner Oberfläche mehrere Stellen, die genau zur dreidimensionalen Struktur des tRNA-Moleküls passen. Die komplexe Struktur des Ribosoms ermöglicht, dass es die mRNA und die beladenen tRNAs in den richtigen Positionen festhalten kann, sodass eine Polypeptidkette effizient zusammengebaut wird. Ein Ribosom kann jede beliebige mRNA und alle Typen von beladenen tRNAs verwenden und eignet sich daher zur Herstellung von Polypeptiden aller Art. Ribosomen können immer wieder verwendet werden, und in den meisten Zelltypen gibt es Tausende davon.
Ein Ribosom besteht aus zwei Untereinheiten, einer großen und einer kleinen. Diese beiden Untereinheiten und mehrere Dutzend weitere Moleküle interagieren nichtkovalent (Abb. 11).
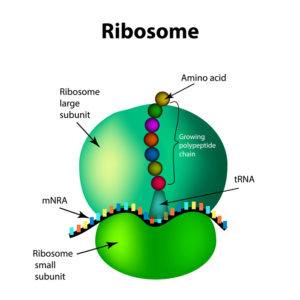
Abb. 11: Ribosom
Bei den Eukaryoten setzt sich die große Untereinheit aus drei verschiedenen Molekülen ribosomaler RNA (rRNA) und 49 verschiedenen Proteinmolekülen zusammen, die in einer ganz bestimmten Weise angeordnet sind. Die kleine Untereinheit umfasst ein rRNA-Molekül und 33 verschiedene Proteinmoleküle.
Die Ribosomen der Prokaryoten sind etwas kleiner als die der Eukaryoten, und ihre Proteine und rRNAs unterscheiden sich etwas von denen der Eukaryoten.
Auf der großen ribosomalen Untereinheit befinden sich drei Stellen, an die eine tRNA binden kann. Sie werden mit A, P und E bezeichnet (Abb. 12). Die mRNA und das Ribosom bewegen sich relativ zueinander, und währenddessen durchwandert die beladene tRNA diese drei Stellen in einer bestimmten Reihenfolge:
![]()
Abb. 12: A-, P-, E-Stelle des Ribosoms
- An der A-(Aminoacyl-)Stelle bindet das Anticodon der beladenen tRNA an das mRNA-Codon und bringt so die richtige Aminosäure an die wachsende Polypeptidkette.
- An der P-(Peptidyl-)Stelle hängt die tRNA ihre Aminosäure an die Polypeptidkette.
- An der E-(Exit-)Stelle befindet sich die tRNA, nachdem sie ihre Aminosäure abgegeben hat, bevor sie vom Ribosom freigesetzt wird und in das Cytosol zurückkehrt. Dort nimmt sie dann eine neue Aminosäure auf und der Vorgang beginnt von vorne.
Das Ribosom besitzt eine Genauigkeitsfunktion, die sicherstellt, dass die Wechselwirkungen zwischen tRNA und mRNA korrekt erfolgen. Das heißt, dass eine beladene tRNA äußerst zuverlässig mit dem richtigen Anticodon an das passende Codon bindet. Wenn es zu einer ordnungsgemäßen Bindung kommt, bilden sich zwischen den Basenpaaren Wasserstoffbrücken. Die rRNA der kleinen Untereinheit ist daran beteiligt, das Zusammenpassen der drei Basen zu überprüfen. Wenn sich nicht zwischen allen drei Basenpaaren Wasserstoffbrücken gebildet haben, muss die tRNA für dieses mRNA-Codon falsch sein und wird aus dem Ribosom entfernt.
Die Translation erfolgt in drei Schritten, die man – wie bei der Transkription – als Initiation, Elongation und Termination bezeichnet (Abb. 13).
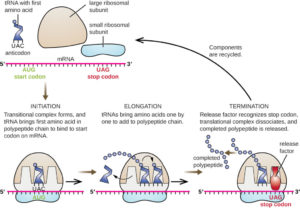
Abb. 13: Translation
Initiation:
Die Translation einer mRNA beginnt mit der Bildung eines Initiationskomplexes, der aus einer beladenen tRNA und einer kleinen ribosomalen Untereinheit besteht, die beide an die mRNA gebunden sind. Die kleine Untereinheit bindet bei Eukaryoten an die 5‘-Cap-Gruppe der mRNA. Bei Prokaryoten bindet die rRNA der kleinen Untereinheit zuerst an eine komplementäre Ribosomenbindungsstelle (AGGAGG,) auf der mRNA. Diese Sequenz liegt weniger als zehn Nucleotide stromaufwärts des eigentlichen Startcodons und ordnet das Startcodon so an, dass es in der Nähe der P-Stelle der großen Untereinheit liegt.
Nach der Bindung wandert die kleine Untereinheit die mRNA entlang, bis sie auf das Startcodon trifft. Das Startcodon hat die Sequenz AUG und die dazu passende tRNA hat die Aminosäure Methionin im Schlepptau. Die erste Aminosäure eines sich bildenden Polypeptids ist also immer ein Methionin. Nach der Bindung der mit Methionin beladenen tRNA an die mRNA tritt die große ribosomale Untereinheit hinzu. Die mit Methionin beladene tRNA befindet sich nun in der P-Stelle des Ribosoms, und in der A-Stelle liegt das zweite mRNA-Codon.
Elongation:
Eine beladene tRNA, deren Anticodon zum zweiten Codon der mRNA komplementär ist, tritt nun in die offene A-Stelle der großen ribosomalen Untereinheit ein. Dann katalysiert die große Untereinheit zwei Reaktionen:
- Die Bindung zwischen der tRNA und ihrer Aminosäure in der P-Stelle wird gelöst.
- Zwischen dieser Aminosäure und derjenigen, die an die in der A-Stelle befindliche tRNA gebunden ist, wird eine Peptidbindung gebildet.
Da die große ribosomale Untereinheit also einen Peptidrest auf eine Aminosäure überträgt (transferiert), spricht man hier von einer Peptidyltransferaseaktivität. Katalysator dieser Reaktion ist die rRNA, weil Experimente, bei denen die Proteinkomponenten der Ribosomen entfernt wurden, nicht jedoch die rRNA, zeigten, dass die Peptidbindung trotzdem weiter stattfand. Die rRNA fungiert also als Ribozym. Nachdem die Peptidbindung zwischen Aminosäure eins und zwei an der A-Stelle geknüpft wurde, bewegt sich das Ribosom entlang der mRNA in 5‘-3‘-Richtung um ein Codon weiter. Die tRNA mit dem Peptidylrest aus zwei Aminosäuren gelangt zur P-Stelle. Die freie tRNA, die mit der ersten Aminosäure beladen war, befindet sich nun an der E-Stelle und verlässt das Ribosom. Sie kann im Cytosol erneut mit Aminosäuren beladen werden. Die A-Stelle ist frei, sodass die nächste tRNA an die mRNA binden kann. Der Vorgang der Elongation setzt sich fort und die Polypeptidkette wächst, während sich diese Schritte wiederholen.
Termination:
Der Elongationszyklus endet und die Translation wird terminiert, sobald ein Stoppcodon – UAA, UAG oder UGA – in die A-Stelle eintritt. Stoppcodons codieren keine Aminosäuren und binden auch keine tRNA. Sie binden vielmehr einen Freisetzungsfaktor (release-Faktor), der die Bindung zwischen dem entstandenen Polypeptid und der tRNA an der P-Stelle hydrolysiert. Das neu gebildete Polypeptid löst sich nun vom Ribosom. Sein C-Terminus ist die letzte Aminosäure, die an die Kette angehängt wurde. Der N-Terminus ist (zumindest zunächst) ein Methionin (als Folge des AUG-Startcodons).