Die Logik der Erstellung von Stammbäumen
Ein evolutionärer Stammbaum, auch phylogenetischer Baum genannt, ist ein Diagramm, das die Geschichte der Divergenz und des evolutionären Wandels zeigt, die von einer einzigen Vorfahrenlinie zu einer Reihe von Nachkommen führt. Mit anderen Worten, er stellt die verwandtschaftlichen Beziehungen einer Gruppe von Organismen dar, wie sie nach Darwins Theorie der Abstammung mit Modifikation von gemeinsamen Vorfahren verstanden werden.
Wir beginnen bei der Erstellung von Stammbäumen mit der Betrachtung eines Idealfalls. Anschließend gehen wir auf Komplikationen ein, die in der realen Welt auftreten.
Stellen wir uns vor, dass wir die evolutionären Beziehungen zwischen den vier fiktiven Vogelarten ableiten wollen, die wir in Abb. 1 sehen.
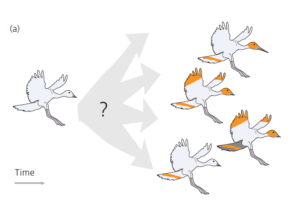
Abb. 1
Text als pdf
Außerdem haben wir folgende Informationen als Voraussetzung:
– Die vier Arten stammen von dem gemeinsamen Vorfahren auf der linken Seite dieser Abbildung ab. Man beachte, dass dieser Vorfahre ein Vogel mit einem kurzen Schnabel mit einem weißen, musterlosen Gefieder ist. Die Kenntnis der Merkmale des gemeinsamen Vorfahren ermöglicht es uns, den langen Schnabel und die verschiedenen Verzierungen, die die vier Arten zieren, als evolutionäre Neuerungen dieser Vogelgruppe zu identifizieren.
– Jede der evolutionären Neuerungen hat sich genau einmal entwickelt. Dieses Wissen erlaubt es uns, die Neuerungen als Indiz für eine gemeinsame Abstammung zu interpretieren.
– Sobald sich jede der Neuerungen in einer Abstammungslinie entwickelt hat, ging sie nicht mehr verloren.
Unter diesen Umständen ist der Rückschluss auf die Evolutionsgeschichte der vier Arten einfach (siehe Felsenstein 1982).
Zunächst wird festgestellt, welche der evolutionären Neuerungen nur bei einer Art vorkommen und welche gemeinsam genutzt werden (Abb. 2). Die dargestellten Merkmale sind: orangener Streifen am Schwanz, gelber Kopf, orangene Flügelspitzen, langer Schnabel und dunkle Schwanzfedern. Die Merkmale, die nur bei einer Art vorkommen – in unserem Fall der lange Schnabel und der dunkle Schwanz – müssen sich entwickelt haben, nachdem sich die Linien, die zu diesen Arten führten, von den Linien, die zu den anderen Arten führten, getrennt haben. Andernfalls würden auch andere Arten lange Schnäbel und dunkle Schwänze haben. Diese Schlussfolgerung ermöglicht es uns, den Stammbaum der Vögel zu zeichnen.
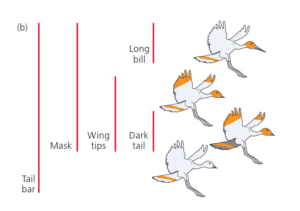
Abb. 2
Wie in Abb. 3 dargestellt, können wir einen Zweig hinzufügen, der zum Vogel mit dem langen Schnabel führt, und einen Zweig, der zum Vogel mit dem dunklen Schwanz führt, und wir können jeden Zweig mit einem Übergang markieren, der das Auftreten des einzigartigen neuen Merkmals anzeigt.
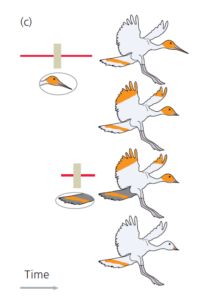
Abb. 3
Nun schauen wir uns die gemeinsamen abgeleiteten Merkmale an. Man beachte, dass orangefarbene Flügelspitzen von genau zwei Arten geteilt werden. Dies weist die Vögel mit orangefarbenen Flügelspitzen als Schwesterarten aus und sagt uns, dass sie einen gemeinsamen Vorfahren haben. Andernfalls würden auch andere Arten orangefarbene Spitzen haben.
Wenn wir unseren Baum, wie Abb. 4 zeigt, in der Zeit zurückverfolgen, können wir nun einen Zweig hinzufügen, der zu der Art mit dem hellen Schwanz und der orangefarbenen Spitze führt, und ihn mit dem Zweig verbinden der zu der orangefarbenen Art mit dunklem Schwanz führt.
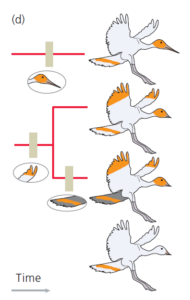
Abb. 4
Der gemeinsame Vorfahre dieser beiden Arten entwickelte die orangefarbenen Flügelspitzen vor der Aufspaltung in diese beiden Arten, entsprechend wird das Merkmal auf diese Stelle eingetragen.
Die beiden Arten mit den orangenen Flügelspitzen und der Vogel mit dem langen Schnabel haben als gemeinsames Merkmal den gelben Kopf, weswegen wir davon ausgehen, dass diese drei Arten enger miteinander verwandt sind als mit der übrig gebliebenen Art, weswegen wir diese Zweige miteinander verbinden. Der gemeinsame Vorfahre dieser drei Arten entwickelte als Merkmal den gelben Kopf.
Alle vier Arten haben als gemeinsames Merkmal den gelben Streifen am Schwanz und das sagt uns, auch, dass sich dieses Merkmal entwickelt haben muss, bevor sich eine der Linien, die zu den vier Vögel führten, voneinander trennen. Wir können nun unseren Baum vervollständigen. Die fertige Phylogenie ist in Abbildung 5 dargestellt.
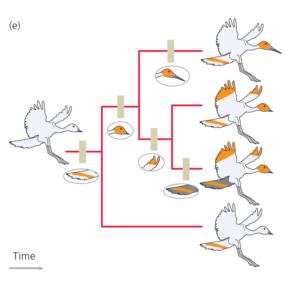
Abb. 5
Wir haben schon im vorherigen Tutorial festgestellt, dass die Abstammung mit Modifikation von einem gemeinsamen Vorfahren automatisch Arten hervorbringt, die verschachtelte Kombinationen gemeinsamer evolutionären Neuerungen aufweisen, und dass die Reihenfolge der Verschachtelung uns erlaubt, die Reihenfolge vorherzusagen in der sich die Neuerungen entwickelt haben und dass wir Darwins Theorie der Abstammung mit Modifikation überprüfen können, indem wir solche Vorhersagen mit dem Fossilbericht vergleichen. Hier haben wir diese Logik noch einen Schritt weitergeführt. Wir haben dieses Prinzip verwendet, um die Geschichte der Diversifizierung zwischen den Abstammungslinien zu rekonstruieren. Das heißt, wir haben gemeinsame abgeleitete Merkmale verwendet, um eine Phylogenie abzuleiten.
Eine evolutionäre Neuheit oder ein abgeleitetes Merkmal wird als Apomorphie bezeichnet. Dies steht im Gegensatz zu einem bereits vorhandenen, ursprünglichen Merkmal, das auch als Plesiomorphie bezeichnet wird. Die richtige Anwendung dieser Begriffe hängt vom jeweiligen Kontext ab.
Betrachten wir hierzu das Merkmal des gelben Kopfes (Abb. 6). Wenn wir alle vier dargestellten Arten berücksichtigen, ist der gelbe Kopf eine Apomorphie, weil drei Arten dieses Merkmal haben, die vierte jedoch nicht. Ein abgeleitetes Merkmal, das von zwei oder mehr oder mehreren Linien geteilt wird, wie z. B. der gelbe Kopf, die von drei Linien innerhalb der Menge der vier lebenden Vogelarten teilen, wird als Synapomorphie bezeichnet. Berücksichtigen wir jedoch nur die drei gelbköpfigen Arten (und ignorieren die vierte Art), so ist dieses Merkmal für die drei gelbköpfigen Arten eine Plesiomorphie, weil sich diese drei Arten eben durch andere Merkmale voneinander unterscheiden (Schnabellänge, Flügelspitzen, Schwanzgefieder).
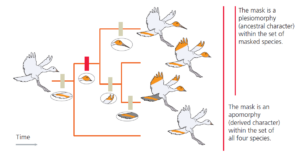
Abb. 6: Ein Merkmal kann in einem Kontext eine Plesiomorphie und in einem anderen eine Apomorphie sein.
Ein weiteres wichtiges Konzept ist das der Monophylie.
Eine monophyletische Gruppe, die auch als Klade bezeichnet wird, besteht aus einem Vorfahren und allen seinen Nachkommen. Bei unseren Vögeln bilden die Arten mit orangefarbener Flügelspitze und ihr gemeinsamer Vorfahre mit diesem Merkmal eine monophyletische Gruppe (Abb. 7a). Diese Gruppe ist in eine andere monophyletische Gruppe eingebettet, die aus den gelbköpfigen Vögeln und ihrem gemeinsamen Vorfahren besteht (Abb. 7b). Und diese Gruppe wiederum ist verschachtelt innerhalb einer anderen monophyletischen Gruppe, die aus den Vögeln mit dem orangen Streifen am Schwanz und ihrem gemeinsamen Vorfahren besteht (Abb. 7c). Eine Gruppe, die aus einem Vorfahren und einigen, aber nicht allen Nachkommen besteht, wie z. B. die hellschwänzigen Vögel und ihr hellschwänziger gemeinsamen Vorfahren in Abbildung 7c, wird als paraphyletisch bezeichnet.
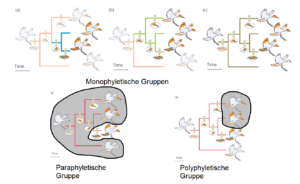
Abb. 7
Eine Gruppe die einige, aber nicht alle Nachkommen eines Vorfahren enthält, und die auch den Vorfahren ausschließt, wie z. B. die Arten mit gelbem Kopf und hellen Schwänzen, wird als polyphyletisch bezeichnet. Wir können nun kurz und bündig ein grundlegendes Prinzip der Phylogenie-Schlussfolgerung darlegen, das von dem deutschen Entomologen Willi Hennig (1966) vertreten wird: Synapomorphien kennzeichnen monophyletische Gruppen.
Phylogenie-Inferenz in nicht idealen Fällen
Unser vorheriges Beispiel zeigte eine recht einfache Phylogenie unter Idealbedingungen, bei dem wir u. a. auch den gemeinsamen Vorfahren der vier Vogelarten kannten.
In den meisten Fällen aber, in denen wir versuchen, die Evolutionsgeschichte zu rekonstruieren, fehlen uns alle besonderen Bedingungen, die für den zuvor betrachteten Idealfall gelten. Erstens kennen wir die Merkmale des gemeinsamen Vorfahren nicht, von dem die betreffenden Arten abstammen. Zweitens entwickeln sich ähnliche evolutionäre Neuerungen manchmal unabhängig voneinander in verschiedenen Abstammungslinien. Und drittens gehen evolutionäre Neuerungen, sobald sie sich entwickelt haben, manchmal verloren.
In diesem Tutorial wollen wir diese Probleme und ihre Lösung veranschaulichen, indem wir drei imaginäre Antilopenarten verwenden, die von einem gemeinsamen Vorfahren abstammen.
War der vorherige Stammbaum eher für Anfänger gedacht, sind wir hier im Fortgeschrittenenkurs.
Abb. 8 zeigt die Antilopen, wobei die interessanten Merkmale hervorgehoben sind.
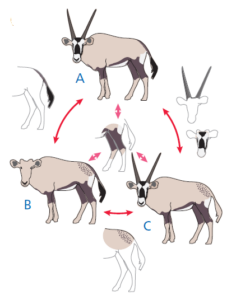
Abb. 8
Wir möchten auf die evolutionären Beziehungen der Antilopen schließen. D.h., welche beiden Arten sind enger miteinander verwandt als eine der beiden mit der dritten? Da wir nicht wissen, wie der jüngste gemeinsame Vorfahre der Antilopen aussieht, wissen wir nicht, welche der Merkmale, die sich zwischen den Arten unterscheiden vom Vorfahren abstammen und welche abgeleitet sind.
Wie Abb. 8 zeigt, ist der Versuch, die Arten nach den gemeinsamen Merkmalen zu gruppieren, wenig sinnvoll. Alle haben braune Beine, aber ansonsten bilden die Arten keine verschachtelten Gruppen. Stattdessen bilden sie Gruppen, die sich willkürlich überschneiden. A und B haben einen dunklen Schwanz; B und C haben einen gefleckten Bürzel; A und C haben Hörner und Masken. Dies deutet darauf hin, dass sich entweder einige der Merkmale mehr als einmal unabhängig voneinander entwickelt haben oder dass einige in den Linien, deren Vorfahren sie hatten, verloren gegangen sind, oder beides. Wie sollen wir da vorgehen?
Hier gibt es zwei Strategien, die uns helfen können, die evolutionäre Vergangenheit der Antilopen zu entwirren: die Outgroup-Analyse und die Parsimony-Analyse vorgestellt.
Bei der Outgroup-Analyse werden eine oder mehrere zusätzliche Arten in unsere historische Rekonstruktion einbezogen (Maddison et al. 1984). Diese sollten mit der Ingroup – den Arten, deren Beziehungen wir ableiten wollen – verwandt sein, aber weniger eng mit ihnen verwandt als die Mitglieder der Ingroup untereinander. Damit ist von vornherein sichergestellt, dass die Ingroup in unserer fertigen Rekonstruktion monophyletisch ist.
In der einfachsten möglichen Outgroup-Analyse, die hier dargestellt ist, fügen wir eine Outgroup mit nur einer Art hinzu (Abb. 9).
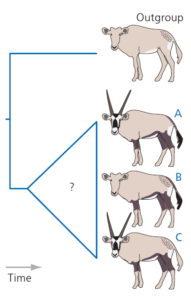
Abb. 9
Wir gehen davon aus, dass es in der Abstammungslinie der Außengruppe keine evolutionären Veränderungen gegeben hat, seit sie sich von der Abstammungslinie, aus der die Innengruppe hervorgegangen ist, getrennt hat. Wie wir gleich sehen werden, erlaubt uns diese Annahme, Rückschlüsse auf die Merkmale des jüngsten gemeinsamen Vorfahren der Ingroup zu ziehen. Es gibt drei mögliche Auflösungen für die Beziehungen zwischen den drei Arten in unserer Ingroup. Entweder sind A und B Schwesterarten oder A und C bzw. B und C. Wir nehmen diese Hypothesen an und vergleichen sie mit Hilfe der Parsimonie-Analyse.
Bei der Parsimony-Analyse bevorzugen wir die Hypothese, die die wenigsten evolutionären Veränderungen bei den interessierenden Merkmalen erfordert (siehe Felsenstein 2004). Wir bewerten jedes Merkmal in jedem möglichen Baum und suchen nach dem einfachsten evolutionären Szenario, das die Verteilung der Merkmalsausprägungen unter den Arten an den Spitzen erklären kann. Wir addieren die Gesamtzahl der evolutionären Veränderungen, die für jede Hypothese und ermitteln die Hypothese mit der geringsten Gesamtzahl.
Die drei möglichen Evolutionsbäume für unsere Antilopenarten sind in Abb. 10 dargestellt. Die einfachsten Szenarien für jedes Merkmal in jedem Baum sind in Abb. 11 dargestellt.
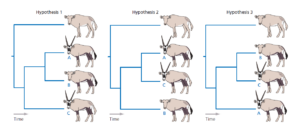
Abb. 10
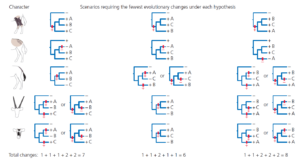
Abb. 11
Wir wollen uns mit den möglichen Szenarien für jedes Merkmal auseinandersetzen.
Braune Beine kommen bei allen drei Antilopenarten unserer Gruppe vor, so dass jede unserer drei Hypothesen nur eine einzige evolutionäre Veränderung erfordert: das Auftreten brauner Beine beim letzten gemeinsamen Vorfahren der Gruppe.
Gefleckte Bürzel kommen in der Außengruppe und in allen Arten der Innengruppe vor, so dass alle drei Hypothesen wiederum nur eine evolutionäre Veränderung erfordern: den Verlust der Bürzelflecken in der jüngsten Vorfahrengeneration von Art A.
Weil die braunen Beine und der gefleckte Bürzel unter den sparsamsten Bedingungen die gleiche Anzahl von Veränderungen erfordern, sind diese für unsere Analyse nicht informativ. Die verbliebenen Merkmale sind hingegen informativ:
Die Verteilung der dunklen Schwänze lässt sich unter Hypothese 1 durch eine einzige evolutionäre Veränderung erklären: ein Zuwachs beim letzten gemeinsamen Vorfahren der Arten A und B. Unter den Hypothesen 2 und 3 sind jedoch zwei Veränderungen erforderlich. Dabei könnte es sich um unabhängiges Auftreten des Merkmals in dem jüngsten Vorfahren von Art A und Art B sein, oder ein Auftreten beim letzten gemeinsamen Vorfahren der Gruppe, gefolgt von einem Verlust in der Linie von Art C.
Die Verteilung der Hörner und Masken kann durch eine einzige Änderung für jedes Merkmal unter Hypothese 2 erklärt werden. Bei den Hypothesen 1 und 3 sind zwei Änderungen für jedes Merkmal erforderlich.
Zählt man die Mindestanzahl der Gesamtveränderungen für jede Hypothese, so stellt man fest Hypothese 1 erfordert sieben Änderungen, Hypothese 2 erfordert sechs und Hypothese 3 acht. Die Parsimony-Analyse veranlasst uns daher, Hypothese 2 als unsere Schätzung der evolutionären Beziehungen zwischen unseren Antilopen der Ingroup zu bevorzugen.
Wie in Abb. 12 gezeigt, können wir auch ableiten, dass der jüngste gemeinsame Vorfahre der Ingroup braune Beine und einen gefleckten Rumpf hatte, dass Hörner und Masken Synapomorphien der Arten A und C sind und dass ein fleckenloser Rumpf eine Apomorphie der Art A ist.
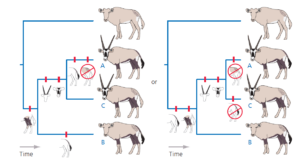
Abb. 12: Phylogenie der Antilopen, abgeleitet aus der Parsimony-Analyse. Diese beiden Hypothesen erfordern die wenigsten evolutionären Veränderungen.
Es könnte verlockend sein, weiter zu folgern, dass der letzte gemeinsame Vorfahre aller vier Arten genauso aussah wie die Außengruppe. Erinnern wir uns jedoch daran, dass wir diese Ähnlichkeit von Anfang an angenommen haben. Ein ebenso plausibles Szenario ist, dass der letzte gemeinsame Vorfahre aller vier Arten braune Beine hatte und dass dieses Merkmal später in der Linie, die zur Außengruppe führte, verloren ging.
Die Parsimony-Analyse ermöglicht es uns, Rückschlüsse auf die Antworten auf Fragen zu ziehen die sonst unlösbar wären. In der Realität verläuft die Evolutionsgeschichte natürlich nicht immer nach dem einfachsten möglichen Szenario ab. Ob die Parsimonie-Analyse gerechtfertigt ist, und aus welchen Gründen, liegt weitgehend außerhalb des Rahmens unseres Tutorials. Einige Biologen halten sie aufgrund von Ockhams Rasiermesser für gerechtfertigt, dem Grundsatz, der besagt, dass einfachere Erklärungen besser sind, wenn alles andere gleichbleibt. Wieder andere sehen sie als gerechtfertigt an, weil sie, wenn sie auf bekannte Evolutionsbäume angewendet wird, einigermaßen genaue Rekonstruktionen liefert.
In unserem Antilopenbeispiel haben wir die Beziehungen zwischen den Arten abgeleitet, indem wir alle möglichen Bäume in Betracht gezogen und denjenigen ausgewählt haben, der die wenigsten evolutionären Veränderungen erfordert. Da unsere Gruppe nur drei Arten umfasste, gab es nur drei mögliche Bäume zu berücksichtigen. Die Zahl der möglichen Bäume nimmt jedoch mit der Zahl der Arten rasch zu (siehe Felsenstein 2004). Bei vier Arten in der Ingroup gibt es 15 mögliche Verzweigungsbäume. Bei fünf Arten sind es 105. Bei 10 Arten gibt es 34.459.425 – mehr als die Anzahl der Sekunden in einem Jahr. Es sollte klar sein, warum in der Praxis die meisten Parsimonie-Analysen mit Hilfe von Computern durchgeführt werden.
Konvergenz und Umkehrung
Das unabhängige Auftreten ähnlicher abgeleiteter Merkmale in verschiedenen Abstammungslinien wird als konvergente Evolution bezeichnet. Der Verlust von abgeleiteten Merkmalen in einer Linie, der zu einer Rückkehr zum Zustand der Vorfahren führt, wird als Umkehrung bezeichnet. Wie wir am Beispiel der Antilope gesehen haben, führen beide Phänomene zu widersprüchlichen Mustern von gemeinsamen abgeleiteten Merkmalen, die uns bei unseren Bemühungen, die Evolutionsgeschichte zu rekonstruieren, in die Irre führen können. Ähnlichkeit in den Merkmalsausprägungen aufgrund von Konvergenz und/oder Umkehrung wird als Homoplasie bezeichnet. Morphologische Ähnlichkeiten können unabhängig voneinander durch konvergente Evolution entstehen, wenn Linien ähnliche Muster der natürliche Selektion aufgrund ähnlicher Umweltbedingungen erfahren. Sowohl Pfaue (Abb. 13a) und männliche Pfauenspinnen (Abb. 13b) tragen bunte Fächer auf dem Rücken und erheben sie, um Partner anzulocken (Hill 2009; Otto und Hill 2010; Dakin & Montgomerie 2011). Sowohl Kaimane (Abb. 13c) als auch Flusspferde (Abb. 13d) haben ihre Augen, Nasenlöcher und Ohren an der Oberseite ihres Schädels. Es wird angenommen, dass dieses Merkmal adaptiv ist, da es ihnen ermöglicht, über dem Wasser zu sehen, zu riechen und zu hören, während sie größtenteils untergetaucht bleiben (Osburn 1903; Caldicott et al. 2005).

Abb. 13: Beispiele für konvergente Evolution
Andere Beispiele der konvergenten Evolution sind die Flügel von Fledermäusen und Vögeln, die stromlinienförmigen Körperformen von Haien und Walen sowie die langgestreckten Körper von Schlangen und beinlosen Eidechsen. Eine Umkehrung kann auftreten, wenn ehemals adaptive abgeleitete Merkmale verloren gehen, weil Umweltveränderungen sie kostspieliger als nützlich gemacht haben (Fong et al. 1995; Hall & Colegrave 2008). Schlangen und beinlose Eidechsen veranschaulichen mit ihrem (konvergenten) Verlust von Gliedmaßen die Umkehrung (siehe Skinner et al. 2008; Skinner & Lee 2009). Andere Umkehrungen umfassen den Verlust der Augen bei höhlenbewohnenden Tieren oder der Zähne bei Vögeln. In einer seltenen doppelten Umkehrung entwickelte die Linie, die zum Guenther-Beutelfrosch (Gastrotheca guentheri) führte, wieder echte Zähne im Unterkiefer – ein Merkmal, das beim gemeinsamen Vorfahren aller modernen Frösche vor mindestens 230 Millionen Jahren verloren ging (Wiens 2011, Abb. 14).

Abb. 14: Guenther-Beutelfrosch (Gastrotheca guentheri)
Wir können uns bei der Schätzung von Phylogenien Ärger ersparen, wenn wir Beispiele für Konvergenz und Umkehrung im Voraus erkennen und dadurch scheinbare Konflikte zwischen den Mustern gemeinsamer abgeleiteter Merkmale in unseren Datensätzen vermeiden können. Mit anderen Worten, es ist sinnvoll, unsere Analysen auf evolutionäre Innovationen zu beschränken, die gemeinsam sind, weil sie homolog sind – von einem gemeinsamen Vorfahren vererbt – und die in allen Nachkommen des gemeinsamen Vorfahren vorhanden sind. Merkmale, die diese Kriterien erfüllen, sind Synapomorphien, die monophyletische Gruppen identifizieren. In einigen Fällen sind Konvergenz und Umkehrung leicht zu erkennen. Niemand würde zum Beispiel die Paarungsrituale des Pfaus und der Pfauenspinne als Beweis für eine gemeinsame Abstammung ansehen. Auch der Verlust von Beinen bei Schlangen lässt sich leicht feststellen, wenn man die rudimentären Gliedmaßen, die bei einigen Arten noch vorhanden sind, und die Existenz fossiler Schlangen mit Beinen betrachtet (Tchernov et al. 2000; Rieppel et al. 2003).
In anderen Fällen wird die Homoplasie nur durch sorgfältige Untersuchungen aufgedeckt. Der prächtige Laubfrosch (Litoria splendida) und der afrikanische Krallenfrosch (Xenopus laevis) scheiden identische Versionen des Hautgiftes Caerulein aus, ein kurzes Protein, das aus 10 Aminosäuren besteht (Abb. 15). Die Produktion von Caerulein könnte leicht als Synapomorphie angesehen werden. Allerdings wird das Protein bei jeder Art von einem anderen Gen kodiert (Roelants et al. 2010). Beim Laubfrosch wird Caerulein von einem Locus kodiert, der aus einer Duplikation des Gastrin-Gens hervorgegangen ist. Beim Krallenfrosch wird Caerulein von einem Locus kodiert, der aus einer Duplikation des Cholecystokinin-Gens hervorgegangen ist. Die Produktion desselben Toxins bei diesen Fröschen ist keine Synapomorphie, sondern ein verblüffendes Beispiel für Konvergenz.
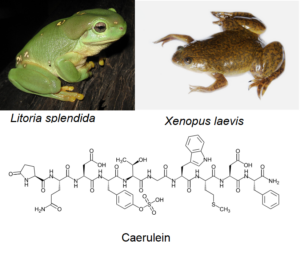
Abb. 15
In anderen Fällen kann Homoplasie nur durch die Rekonstruktion einer Phylogenie entdeckt werden, wobei festgestellt wird, dass die Verteilung einiger Merkmale nicht ohne Konvergenz erklärt werden kann (Wake et al. 2011).
Mit anderen Worten: Homoplasie ist eine Tatsache in der Phylogenie. Sie stellt das „Rauschen“ in den Datensätzen, die zur Rekonstruktion der Evolutionsgeschichte verwendet werden. Der beste Weg, um zu vermeiden, dass man durch das Rauschen der Homoplasie in die Irre geführt wird, ist die Analyse vieler statt nur einiger unabhängiger Merkmale bei der Rekonstruktion der Beziehungen. Mooi et al. (2000) verwendeten eine Parsimonie-Analyse von 24 strukturellen Merkmalen, um den evolutionären Baum der fossilen Sanddollars zu schätzen, der in 16 dargestellt ist. Der sparsamste Baum erforderte nur jeweils einen Übergang für 21 der Merkmale. Diese Übergänge sind blau markiert. Die verbleibenden drei Merkmale, die durch andere Farben gekennzeichnet sind, waren homoplastisch. Jedes von ihnen erforderte zwei Zustandsänderungen.
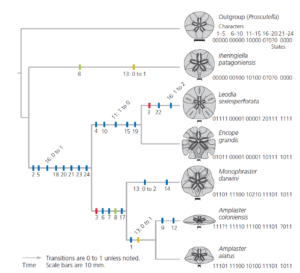
Abb.16: Phylogenie der fossilen Sanddollars durch Parsimony-Analyse geschätzt. Für die meisten anatomischen Merkmale, die zur Ableitung dieses Stammbaums verwendet wurden, gibt es zwei mögliche Zustände; einige wenige haben drei. Die Zahlenreihe neben jedem Sanddollar gibt den Zustand für jedes Merkmal an. Nachgezeichnet aus Mooi et al. (2000).
In diesem Tutorial haben wir uns mit der Rekonstruktion des Stammbaums von drei fiktiven Antilopenarten mithilfe des Parsimonie-Verfahren befasst und dazu einige reale Beispiele aus der Praxis gezeigt. Das Parsimonie-Verfahren ist aber längst nicht das einzige Verfahren in der Kladistik. Im Nächsten Video gehen wir in den Experten-Modus über. Bleibt also dran.
Literatur
Caldicott, D. G., D. Croser, et al. (2005): Crocodile attack in Australia: An analysis of its incidence and review of the pathology and management of crocodilian attacks in general. Wilderness & Environmental Medicine 16:143–159.
Dakin, R., and R. Montgomerie (2011) Peahens prefer peacocks displaying more eyespots, but rarely. Animal Behaviour 82:21–28.
Felsenstein, J. (1982): Numerical methods for inferring evolutionary trees. Quarterly Review of Biology 57:379–404.
Felsenstein, J. (2004): Inferring Phylogenies. Sunderland, MA: Sinauer
Fong, D. W., T. C. Kane, and D. C. Culver (1995): Vestigialization and loss of nonfunctional characters. Annual Review of Ecology and Systematics 26:249–268.
Hall, A. R., and N. Colegrave (2008): Decay of unused characters by selection and drift. Journal of Evolutionary Biology 21:610–617.
Hennig, W. (1966): Phylogenetic Systematics. Translated by D. D. Davis and R. Zangerl. Urbana: University of Illinois Press.
Hill, D. E. (2009): Euophryine jumping spiders that extend their third legs during courtship (Araneae: Salticidae: Euophryinae: Maratus, Saitis). Peckhamia 74.1:1–27
Maddison, W. P., M. J. Donoghue, and D. R. Maddison (1984): Outgroup analysis and parsimony. Systematic Zoology 33:83–103.
Mooi, R., S. Martinez, and S. G. Parma (2000): Phylogenetic systematics of Tertiary monophorasterid sand dollars (Clypeasteroida: Echinoidea) from South America. Journal of Paleontology 74:263–281.
Osburn, R. C. (1903): Adaptation to aquatic, arboreal, fossorial and cursorial habits in mammals. I. Aquatic adaptations. American Naturalist 37:651–665.
Rieppel, O., H. Zaher, et al. (2003): The anatomy and relationships of Haasiophis terrasanctus, a fossil snake with well-developed hind limbs from the mid-cretaceous of the Middle East. Journal of Paleontology 77:536–558.
Roelants, K., B. G. Fry, et al. (2010): Identical skin toxins by convergent molecular adaptation in frogs. Current Biology 20:125–130.
Skinner, A., and M. S. Y. Lee (2009): Body-form evolution in the scincid lizard clade Lerista and the mode of macroevolutionary transitions. Evolutionary Biology 36:292–300.
Skinner, A., M. S. Y. Lee, and M. N. Hutchinson (2008): Rapid and repeated limb loss in a clade of scincid lizards. BMC Evolutionary Biology 8: 310.
Tchernov, E., O. Rieppel, et al. (2000): A fossil snake with limbs. Science 287:2010–2012.
Wake, D. B., M. H. Wake, and C. D. Specht (2011): Homoplasy: From detecting pattern to determining process and mechanism of evolution. Science 331:1032–1035.
Wiens, J. J. (2011): Re-evolution of lost mandibular teeth in frogs after more than 200 million years, and re-evaluating Dollo’s law. Evolution 65:1283–1296.