Isomerie
Bei vielen organischen Molekülen findet sich das Phänomen der Isomerie. Isomerie bedeutet, dass die Summenformel des Moleküls die gleiche ist, die Strukturformel jedoch eine andere. Das heißt, dass ein Molekül die gleiche Anzahl von Atomen haben kann, diese aber unterschiedlich angeordnet sein können.
Sehen wir uns hierfür als Beispiel das Propanol an (Abb.1). Propanol gehört zur Familie der Alkohole, welche durch eine OH-Gruppe als funktionelle Gruppe gekennzeichnet sind. Propanol ist ein Alkohol mit drei Kohlenstoffatomen. Beim Propanol kann die OH-Gruppe entweder an einem der äußeren Kohlenstoffatome verbunden sein, dann bezeichnet man diese Form des Propanols als Propan-1-ol (bzw. n-Propanol). Die OH-Gruppe kann aber auch am mittleren Kohlenstoffatom verbunden sein. Die Zahl der Atome, also die Summenformel, ist die gleiche (C3H8O; bzw. auch als C3H7OH). Die Position der funktionellen Gruppe jedoch eine andere, womit wir eine andere Strukturformel haben. Dieses Isomer des Propanols bezeichnet man als Propan-2-ol. Solche Strukturisomere haben dann auch andere chemische und physikalische Eigenschaften.
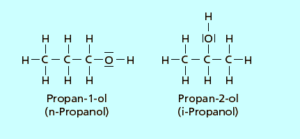
Abb. 1: Isomere von Propanol
Chiralität
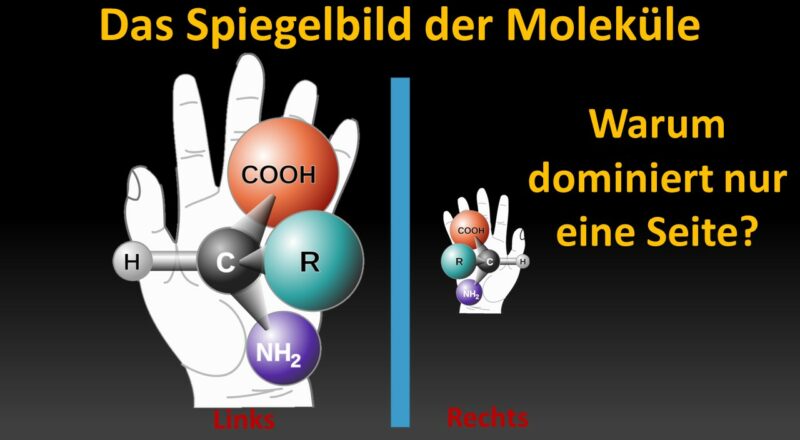
Neben diesen Strukturisomeren kommt bei organischen Molekülen auch etwas vor, dass man als Chiralität bezeichnet (Abb. 2).
Text als pdf
Der Begriff Chiralität („Händigkeit“) betrifft die Struktur zweier Molekülsorten, die wie Bild und Spiegelbild gebaut sind und somit nicht durch Drehung zur Deckung gebracht werden können. Dies kann durch die atomare Anordnung im Molekül oder durch die dreidimensionale Gestalt der Moleküle selbst bedingt sein. So werden chirale Zentren im Molekül z. B. von Kohlenstoffatomen gebildet, die vier verschiedene Atomgruppen tragen.
Chiralität ist wie mit unseren Händen vergleichbar: dieselbe Anzahl an Fingern, Knochen, Muskeln etc. und doch können wir unsere Hände in eine linke und eine rechte Hand unterscheiden.
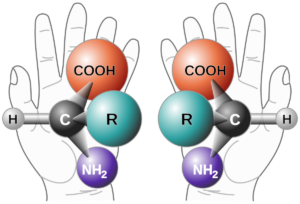
Abb. 2: Grundprinzip der Chiralität
Derart asymmetrisch gebaute Moleküle drehen die Ebene von polarisiertem Licht, das durch eine Lösung oder einen Kristall asymmetrischer Moleküle geschickt wird, nach rechts oder nach links. Man sagt auch, dass solche Substanzen „optisch aktiv“ seien (Abb. 3). Moleküle, die sich wie Bild und Spiegelbild verhalten, werden als Enantiomere bezeichnet; ihr chemisches und physikalisches Verhalten ist gegenüber nicht-chiralen Einflüssen, wie z. B. unpolarisiertem Licht oder Reaktionen mit nicht-chiralen Molekülen, tatsächlich gleich. In chiraler Umgebung hingegen, d. h. in polarisiertem Licht, bei Wechselwirkung mit Oberflächen chiraler Struktur oder in der Reaktion mit anderen chiralen Molekülen, sind Enantiomere physikalisch und chemisch deutlich voneinander unterscheidbar (Mortimer & Müller 2019, Rein 1993).
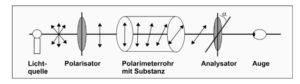
Abb. 3: Drehung der Ebene polarisierten Lichts, das durch einen Polarisator im Polarimeterrohr erzeugt wird, durch die jeweilige Substanz, die sich normalerweise in einer wässrigen Lösung befindet. Mit dem Analysator kann man den Drehwinkel der Schwingungsebene des Lichts messen.
Im Rahmen klassischer Synthesen treten stets Gemische unterschiedlicher Enantiomeren auf (so genannte Razemate). Bemerkenswerterweise wird bei Lebewesen meist nur eine der Spiegelbild-Isomerien bevorzugt.; z. B. kommen in der Natur ausschließlich L-Aminosäuren und D-Zucker vor. Mit anderen Worten: Das Leben ist homochiral.
D-Aminosäuren und L-Kohlenhydrate in der Natur
Jedoch gibt es auch hier Ausnahmen von der Regel.
Die Forschung hat gezeigt (Brückner & Fujii 2010), dass D-Aminosäuren in Biosystemen wesentlich weiterverbreitet sind als bisher angenommen und wurden in Pflanzen, Wirbellosen und Wirbeltieren gefunden (Robinson 1976, Corrigan 1969, Aliashkevich et al. 2018, Abb. 4). Später wurden D-Aminosäuren auch in menschlichem Hirngewebe, Zähnen und der Augenlinse entdeckt (Masters 1978).
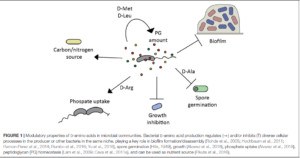
Abb. 4: Modulierende Eigenschaften von D-Aminosäuren in mikrobiellen Gemeinschaften. Die bakterielle D-Aminosäureproduktion reguliert (->) und/oder hemmt (T) verschiedene zelluläre Prozesse im Produzenten oder in anderen Bakterien in derselben Nische und spielt eine Schlüsselrolle bei der Bildung/Demontage von Biofilmen (Rohde et al. 2005; Hochbaum et al. 2011; Ramon-Perez et al. 2014; Rumbo et al. 2016; Yu et al. 2016), der Sporenkeimung (Hills 1949), dem Wachstum (Alvarez et al. 2018), der Phosphataufnahme (Alvarez et al. 2018) und der Peptidoglykan (PG)-Homöostase (Lam et al. 2009; Cava et al. 2011a) und kann als Nährstoffquelle genutzt werden (Pikuta et al. 2016).
D-Aminosäuren finden sich im Gewebe von Wanzen, in der Zellwand von Bakterien (Stevens et al. 1951, Sela & Zisman 1997), im Nervensystem von Kopffüßern und Amphibien (Avnir 1994, Mor et al. 1992), in Spinnengiften (Kreil 1994), im Hautgift einiger südamerikanischer Frösche sowie in vielen bakteriellen Antibiotikaprodukten (Jung 1992).
Die am häufigsten vorkommenden D-Aminosäuren in Wirbeltieren sind D-Aspartat und D-Serin. Es findet sich bei Säugetieren eine beträchtliche Menge an D-Serin ausschließlich im Gehirn mit einer Konzentration von etwa einem Drittel des L-Serins, wobei die höchsten Werte im Vorderhirn zu finden sind (Hashimoto et al. 1993a, b, 1997). D-Aspartat ist in hohem Maße in neuroendokrinen Geweben lokalisiert, so z. B. im Nebennierenmark, in Oxytocin/Vasopressin-Neuronen, in der Zirbeldrüse und in bestimmten Neuronenpopulationen im Gehirn (Schell et al. 1997). Zahlreiche neuere Erkenntnisse sprechen für eine Neurotransmitter-/Neuromodulatorrolle von D-Serin. D-Serin wird aus L-Serin durch Serin-Racemase in den astrozytären Gliazellen synthetisiert, die Synapsen umhüllen, insbesondere in Gehirnregionen, die reich an NMDA (N-Methyl-D-Aspartat-Rezeptor), einem Glutamatrezeptor, sind (Mustafa et al. 2007, Abb. 5).
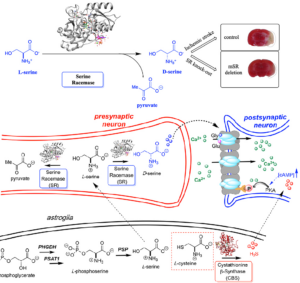
Abb. 5: oben: Auswirkung von D-Ser auf das Infarktvolumen nach einem ischämischen Schlaganfall, der durch einen vorübergehenden Verschluss der mittleren Hirnarterie (tMCAO) ausgelöst wurde – SR-Knockout-Mäuse im Vergleich zur Kontrollgruppe. Unten: L-Serin wird zunächst in den Astroglia aus 3-Phosphoglycerat über eine Kaskade von drei Enzymen hergestellt, an der Phosphoglycerat-Dehydrogenase (PHGDH), Phosphoserin-Aminotransferase 1 (PSAT1) und Phosphoserin-Phosphatase (PSP) beteiligt sind. L-Serin wird dann zum präsynaptischen Neuron transportiert, wo es durch hSR in D-Ser umgewandelt wird. D-Serin wirkt als starker NMDAR-Koagonist an der Glycinstelle. Es wird angenommen, dass das von CBS erzeugte H2S den NMDA-Rezeptor ebenfalls aktiviert. Zu den Modellen für die H2S-NMDAR-Aktivierung gehören eine cAMP-abhängige PKA-vermittelte Phosphorylierung und eine H2S- oder Sulfan-Schwefel-vermittelte NMDAR-Disulfidbindungsreduktion. Nach Graham et al. (2019), Kimura (2000), Kimura et al. (2013) und Mustafa et al. (2010).
In den letzten drei Jahrzehnten hat die Forschung zu D-Aminosäuren beim Menschen deren Häufigkeit im Gehirn sowie in anderen Geweben und Körperflüssigkeiten bestätigt (Bastings et al. 2019).
Es hat sich zudem gezeigt, dass die Menge an D-Aminosäuren mit zunehmendem Alter eines Organismus ansteigt, was zu physiologischen Veränderungen und zu altersbedingten Krankheiten, inklusive Arteriosklerose und Alzheimer führen kann (Fujii et al. 2018, Masters et al. 1977, Fisher et al. 1994, 1995, Nagata et al. 1995, Schell 1995, Konno 2005).
Die D-Aminosäuren im Körper können aus mehreren Quellen stammen. So sind Racemase-Enzyme in der Lage D-Aminosäuren herzustellen. Derzeit sind zwei Racemase-Enzyme in Säugetieren bekannt: Serin-Racemase und Aspartat-Racemase (Ohide et al. 2011). Im menschlichen Gewebe wurde nur die Serin-Racemase gefunden (Wolosker et al. 1999, Foltyn et al. 2005, Raboni et al. 2019). Aspartat-Racemase hingegen wurde bei Mäusen, Amphibien, Mollusken und Bakterien nachgewiesen (Uda et al. 2016).
Zweitens können D-Aminosäuren über die Nahrung in den Körper gelangen. Bestimmte Verfahren der Lebensmittelverarbeitung tragen zur Racemisierung von L-Aminosäuren in D-Aminosäuren bei (Friedman et al. 2010). Darüber hinaus finden sich D-Aminosäuren in fermentierten Lebensmitteln wie Essig und Milchprodukten (Mutaguci et al. 2013, Jin et al. 1999). Drittens wird angenommen, dass mindestens ein Drittel des menschlichen D-Aminosäure-Pools aus der mikrobiellen Synthese, z. B. Darmbakterien, stammt (Foltyn et al. 2005, Aliashkevich et al. 2018, Sasabe et al. 2016).
Obwohl L-Zucker in der Natur äußerst selten vorkommen (Pikuta et al. 2006), stellen einige Ausnahmen wie Arabinose, Fucose und Rhamnose eine Besonderheit unter den einfachen Monosacchariden dar, da sie häufiger in der L- als in der D-Konfiguration auftreten. Darüber hinaus ist L-Galaktose (L-Gal) ein seltener Zucker, der in terrestrischen Organismen als Bestandteil von Polysacchariden in einigen Pflanzen, Schnecken, Rotalgen und Weichkorallen gefunden wurde (Gutiérrez et al. 2004). Darüber hinaus gibt es noch den Extremfall der Rhamnose, die in der Natur nur in ihrer L-Form vorkommt. Sie ist häufig an andere Zucker gebunden, die in Pflanzen und Kieselalgen vorkommen und Bestandteil der äußeren Zellmembranen sind (Abb. 6).
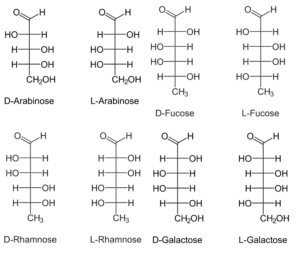
Abb. 6: Einige Zuckerarten, die in ihrer L-Konfiguration häufiger auftreten als in ihrer D-Konfiguration.
Es lohnt sich, einige Referenzen zu erwähnen, die sich mit D-Aminosäuren befassen, insbesondere einige Übersichtsarbeiten zu diesem Thema, z. B. (Devinski 2021, Fujii 2002, Deamer et al. 2007, Brückner & Fujii 2011, Gaspar et al. 2013, Genchi 2017, Pundir et al. 2017, Grishin et al. 2020).
Ursachen der Homochiralität
Dennoch dominieren L-Aminosäuren und D-Kohlenhydrate die belebte Welt. Viele Theorien versuchen, das Phänomen der Homochiralität zu erklären, aber keine hat eine befriedigende Erklärung geliefert (Podlech 2001, Gusev 2001, Plasson et al. 2007).
Warum ist die Grundlage unseres Lebens auf L-Aminosäuren und D-Sacchariden aufgebaut, obwohl theoretisch nichts dagegenspricht, die Biomoleküle aus entgegengesetzten Enantiomeren aufzubauen?
Hat das Leben mit beiden Formen der Chiralität begonnen, und ist dann eine der Formen verschwunden? Oder ging die Wahl der Homochiralität der Bildung von Biomolekülen voraus, die die Replikation und Informationsübertragung gewährleisten konnten? Ist die natürliche Auswahl von L-Aminosäuren und D-Zuckern, auf denen das Leben basiert, deterministisch oder zufällig? Ist die Händigkeit im Universums seit dem Urknall vorhanden (Mauksch et al. 2010, Riehl 2010)? Diesen Geheimnissen versuchen verschiedene Arbeitsgruppen zu lösen.
Wie kam es im Lauf der Evolution zur Diskriminierung eines Enantiomers oder anders gefragt: Wie kam es zu einem Überschuss eines Enantiomers (kurz ee)? Die Idee, dass durch Zufall ein bestimmtes Enantiomer in den ersten präbiotischen Synthesen lokal überwog und anschließend in einer Kettenreaktion andere Moleküle gleicher Chiralität koppelte, entspricht dem Prinzip eines sich selbst aufschaukelnden Automatismus (Fajszi & Czege 1981, Jenkins et al. 1994). Der kleine Vorsprung des „Überlebenden“ würde sich zu Ungunsten des erfolgloseren Enantiomers stets vergrößern. Nur erweisen sich in allen Syntheseversuchen biotischer Moleküle „falsche“ Enantiomere als ebenso reaktionsfreudig und werden ungeachtet ihrer Chiralität in gleichem Maße eingebaut (Bailey 2000, Bonner & Dean 2000).
Dies gilt jedoch nicht notwendigerweise für katalytisch gesteuerte Reaktionen, die in den sehr heterogenen Nischen der Ozeane ablaufen konnten. Es gibt empirische Hinweise, die dafürsprechen, dass z. B. durch Komplexierung (etwa durch Cu2+-Ionen) aus razemischen Aminosäure-Gemischen bevorzugt homochirale Proteine kondensieren (Plankensteiner et al. 2004).
Den möglichen Impuls für eine Veränderung des Enantiomeren-Verhältnisses sind in der Auswirkung chiraler, mineralischer Oberflächen auf die Kristallisation zu suchen. Bekannt dafür sind Quarz, Tonmineralien oder Feldspat (Hazen 2001, Hazen et al. 2001). Auch an reinen Calciten können Enantiomeren-Spezies angereichert und damit Razemate mit hoher Selektivität aufgetrennt werden (Abb. 7).
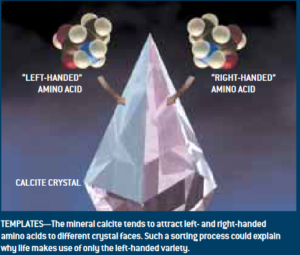
Abb. 7: Das Mineral Calcit neigt dazu, links- und rechtsdrehende Aminosäuren auf unterschiedliche Kristallflächen zu ziehen. Ein solcher Sortierprozess könnte erklären warum das Leben nur die linkshändige Variante nutzt.
Dabei kristallisiert bevorzugt immer nur ein bestimmtes optisches Isomer aus. Des Weiteren lassen sich aus übersättigten Lösungen unter dem Einfluss asymmetrischer Kristallisationskeime entweder D- oder L-Formen auskristallisieren, wobei die bereits auskristallisierte Form als Kristallisationskeim dienen kann (Harada 1970). Die Entstehung übersättigter Lösungen ist in räumlich getrennten Gewässern durch Austrocknung oder über erhitzten Gesteinen geologisch durchaus möglich. Schließlich wird durch polarisierte ß-Strahlung aus dem natürlichen 90Sr-Zerfall das D-Tyrosin stärker zerstört als L-Tyrosin (Follmann 1981).
Auch Meereis hat offenbar chirale Einflüsse (Trinks 2001). So stellt die Bildung von Kristallstrukturen in asymmetrischer, wendelförmiger Anordnung ein bei der Entstehung von Kristallen häufig anzutreffendes Phänomen dar, so auch im Eis (Hillig & Turnbull 1956, Hobbs & Ketcham 1969, Kirkpatrick 1975, Trinks 2001).
Haben wir eine Mischung von zwei Enantiomeren, die in getrennten Kristallen vorliegen, so spricht man von einem Konglomerat (Abb. 8). Die Elementarzellen jedes einzelnen Kristalls bestehen also ausschließlich aus einem der beiden Enantiomeren. Konglomerate können eine spontane Trennung der Enantiomere entwickeln, bei dem eines der Enantiomere kristallisiert und die andere in der Lösung verbleibt.
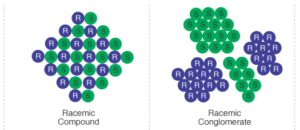
Abb. 8: Abbildung zum Vergleich einer racemischen Verbindung mit einem racemischen Konglomerat.
Die meisten natürlichen Aminosäuren sind bei moderaten Temperaturen jedoch racemische Verbindungen, man hat also eine Mischung aus beiden Enantiomeren. Man konnte jedoch zeigen, dass Racemate mit Asparaginsäure und Glutaminsäure in Gegenwart von Sedimentoberflächen als Konglomerate auskristallisieren können und sich die Enantiomere trennen können (Viedma 2001). Eine asymmetrische Beeinflussung geschieht natürlich bei Zusatz enantiomerenreiner Impfkristalle (Asakura et al. 2000), aber auch spontan und ohne Außeneinwirkungen wie bei der Kristallisation eines Isomers aus einer wässrigen Aminosäurelösung (Shinitzky et al. 2002). Einer Arbeitsgruppe gelang die Synthese von Oligopeptiden aus Serin mit erstaunlicher chiraler Reinheit (Schalley & Weis 2002).
Reiner et al. (2006) konnten sogar nachweisen, dass Die Aminosäure L-Histidin begünstigt die Bildung homochiraler Oligopeptide, so dass sie eine Schlüsselrolle bei der Entstehung homochiraler Proteine gespielt haben könnte.
Mehrere Experimente deuten darauf hin, dass eine Sublimation eines Enantiomerengemisches zu einer Anreicherung der Enantiomere führen kann (Fletscher et al. 2007, Blackmond & Klussmann 2007, Abb. 9). Eine Sublimation stellt einen Phasenübergang von einen festen in einen gasförmigen Zustand dar.
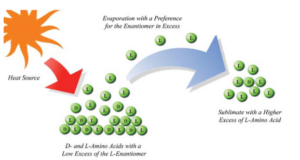
Abb. 9: Anreicherung von Enantiomeren durch Sublimation
So konnte eine Arbeitsgruppe im Jahr 2013 durch Sublimation verschiedener Aminosäure-Racemate eine Anreicherung der einzelnen Enantiomere erreichen (Tarasevych et al. 2013, Abb. 10).
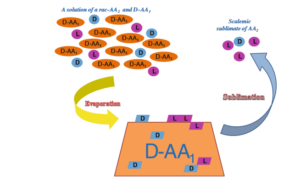
Abb. 10: Sublimation eines gemeinsam ausgefällten Gemischs aus DL-aliphatischem AA 2 und nicht flüchtigem D-AA1
Weiterhin kann die Gluconeogenese in Gegenwart katalytisch aktiver Aminosäuren stereoselektiv ablaufen (Pizzarello & Weber 2004, Weber & Pizzarello 2006). Die Verbindung zwischen den in der Natur überwiegend vorkommenden L-Aminosäuren und den überwiegend natürlich vorkommenden D-Zuckern wurde für Serinoktamere hergestellt (Nanita & Cooks 2006).
Bereits ein 1 %iger Überschuss an L-Isomeren kann durch einfaches Verdampfen einer Lösung zu einem Verhältnis von 95:5 zwischen L und D-Isomeren verstärkt werden, so dass das Leben mit einer solchen Lösung beginnen könnte, in der die dominierenden L-Isomere selektiv ausgewählt werden. Gleichzeitig haben offenbar D-Zucker, die bevorzugten Enantiomere der Kohlenhydrate bei Lebewesen, einen Selektionsvorteil, weil L-Aminosäuren unter präbiotischen Bedingungen die Bildung eines Überschusses an D-Glyceraldehyd und damit an anderen D-Zuckern katalysieren (Breslow & Cheng 2010, Abb. 11).
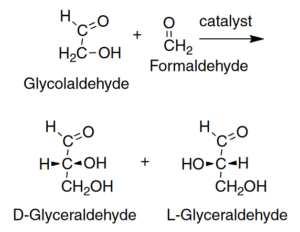
Abb. 11: Die Aldolreaktion zwischen Glykolaldehyd und Formaldehyd kann sowohl zu D-Glyceraldehyd als auch zu L-Glyceraldehyd führen, wobei jedoch das D-Isomer bevorzugt wird, wenn eine L-Aminosäure als Katalysator dient.
Diese Theorie steht im Einklang mit der Arbeit von Morowitz (1969), der den theoretischen Hintergrund solcher Prozesse und einige experimentelle Untersuchungen dazu beschrieben hat. Andererseits gibt es starke Zweifel an dieser Theorie, z. B. (Bada 2009) die die zentrale Idee des Transfers und der Verstärkung des Enantiomerenüberschusses auf andere Moleküle in Frage stellen, so dass das Bild alles andere als eindeutig ist. Solche Spekulationen beruhen auf Laborergebnissen, die die präbiotischen Bedingungen auf der Erde definitiv nicht exakt und vollständig simulieren können.
Hein et al. (2011) berichten über die Synthese von RNA-Vorläufermolekülen, die aus racemischen Ausgangsmaterialien einen Typ von Enantiomeren anreichern, wobei die molekulare Asymmetrie lediglich auf ein geringes anfängliches Ungleichgewicht der in der Reaktionsmischung vorhandenen Aminosäure-Enantiomere zurückzuführen ist (Abb. 12). Die Entstehung von Molekülen mit einer einzigen Chiralität aus komplexen Multikomponenten-Mischungen untermauert die Robustheit dieses Syntheseprozesses unter potenziell präbiotischen Bedingungen und liefert eine plausible Erklärung für die Bevorzugung biologischer Moleküle vor der Entstehung selbstreplizierender Informationspolymere.
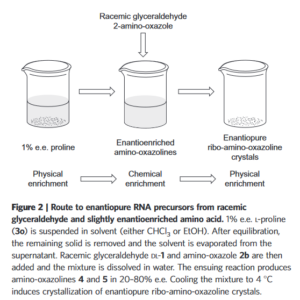
Abb. 12: Weg zu enantioreinen RNA-Vorläufern aus racemischem Glyceraldehyd und leicht enantioreicher Aminosäure. 1% e. e. L-Prolin wird in einem Lösungsmittel (entweder CHCl3 oder EtOH) suspendiert. Nach der Äquilibrierung wird der restliche Feststoff entfernt und das Lösungsmittel aus dem Überstand verdampft. Anschließend werden racemisches Glyceraldehyd DL-1 und Amino-Oxazol zugegeben und das Gemisch in Wasser aufgelöst. Bei der anschließenden Reaktion entstehen Aminooxazoline in 20-80 % e.e. Das Abkühlen der Mischung auf 4 °C führt zur Kristallisation von enantiomerenreinen Ribo-Amino-Oxazolin-Kristallen.
Anhand von Computersimulationen konnte gezeigt werden, dass die einseitige Chiralität auf Polymerebene entstanden sein könnte, und zwar aus einem racemischen Gemisch von Monomeren. Mit anderen Worten, die Ergebnisse deuten darauf hin, dass die Homochiralität mit dem Aufkommen der Biopolymere während der Entstehung des Lebens entstanden sein könnte und nicht auf der Ebene der Monomere vor der Entstehung des Lebens (Chen & Ma 2020, Abb. 13).
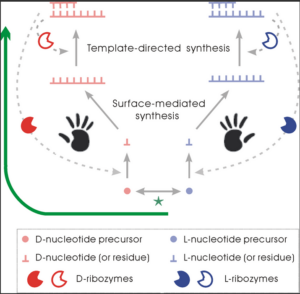
Abb. 13: Die Entstehung der Homochiralität auf der Polymerebene in der RNA-Welt. Die Entwicklung beginnt mit einem racemischen Pool von Nukleotidvorläufern, in dem die beiden chiralen Typen leicht ineinander übergehen können (der grüne Stern). Die Vorliebe der RNA, bei der De-novo-Polymerisation (oberflächenvermittelte Synthese) und der Replikation (Template-gesteuerte Synthese) Monomere mit der gleichen Chiralität wie die eigene einzubauen („chirale Selektion“), führt zu der autokatalytischen Eigenschaft, die den geringfügigen Unterschied zwischen den beiden chiralen Formen verstärken kann – was vielleicht zunächst zufällig auftritt. Während der Verstärkung verschieben sich die Materialien in der entgegengesetzten Form durch den Racemisierungsausgleich (der grüne Stern) in Richtung der Zielform. Infolgedessen kommt es zu einer erheblichen Chiralitätsabweichung des gesamten Systems (grüner Pfeil), wodurch lange RNA-Ketten mit einheitlicher Händigkeit entstehen, die das Auftreten von Ribozymen ermöglichen. Das Aufkommen von Ribozymen (siehe Text für Erklärungen zu solchen Ribozymen) kann die Chiralitätsabweichung aufgrund ihrer spezifischeren, effizienteren chiralen Selektion noch weiter verstärken. Das heißt, die daraus resultierende biologische Chiralität des einen Typs anstelle des anderen Typs sollte ursprünglich zufällig auf diese Weise entstanden sein.
Da diese Biopolymer-Katalysatoren chiral sind, führt derselbe Übergang das System in einen homochiralen Zustand. Homochiralität wird also aufrechterhalten, weil Biopolymere in ihrer Katalyse einen Enantiomertyp bevorzugen und so ihre eigene Chiralität katalyisieren (vgl. auch Wu et al. 2012, Abb. 14).
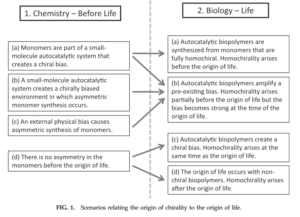
Abb. 14: Szenarien, die den Ursprung der Chiralität mit dem Ursprung des Lebens in Verbindung bringen.
Biopolymere wie z. B. RNA oder Proteine, sind nach den Hypothesen der Vertreter der in der letzten Episode vorgestellten Lipid-Welt-Hypothese erst nach der Bildung supramolekularer Strukturen wie z. B. Vesikel oder Oparins Koazervate entstanden (Deamer 2017, Ikehara 2005). Trifft diese Hypothese zu, dürften, nach der Ansicht einiger Forscher, diese amphiphilen Moleküle eine Rolle bei der Entstehung der biologischen Homochiralität gespielt haben, wenn nicht unbedingt die entscheidende (Suzuki & Itabashi 2019, Abb. 15).
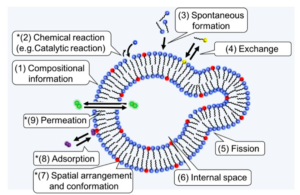
Abb. 15: Funktionen von Amphiphilen, die für den Ursprung des Lebens und die Homochiralität wichtig sein könnten.
Die asymmetrische Autokatalyse ist aber auch mit kleinen Molekülen möglich, wie die Soai-Reaktion zeigt, aber sie ist selten. Die Soai-Reaktion (Soai et al., 1995, 2001, Abb. 16) ist die Reaktion von 5-Pyrimidylalkanol mit Diisopropylzink. Dies ist ein gut dokumentierter Fall eines chemischen Systems mit kleinen Molekülen, der zeigt, dass ein großer Enantiomerenüberschuss durch asymmetrische Autokatalyse entstehen kann. Obwohl dies das Prinzip demonstriert, dass ein homochirales System kleiner Moleküle spontan entstehen kann, sind die spezifischen chemischen Komponenten der Soai-Reaktion für die präbiotische Chemie nicht direkt relevant.
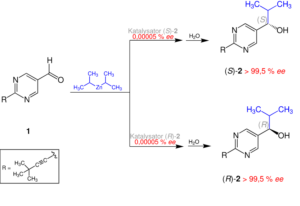
Abb. 16: Soai-Reaktion mit asymmetrischer Autokatalyse und Erhöhung des ee
Physikalische Ursachen
Einige Forscher suchen aber nach der Quelle der Homochiralität nicht bei den Molekülen auf der Erde oder in Meteoriten, sondern schon gleich im Ursprung des Universums.
Eine Ursache könnte dabei mit den Neutrinos zusammenhängen. Machen wir dazu nochmal einen Exkurs zum Standardmodell der Teilchenphysik (Abb. 17)[1].
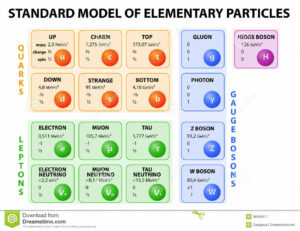
Abb. 17: Standardmodell der Teilchenphysik
Materie besteht aus Materieteilchen (auch Fermionen genannt). Insgesamt gibt es zwölf Materieteilchen, die in sechs Quarks und sechs Leptonen unterteilt werden. Beide Gruppen bestehen aus Teilchen dreier Generationen. Die Quarks der ersten Generation sind die Up- und Down-Quarks. Die zweite Generation besteht aus den Charme- und Strange-Quarks und die dritte aus Top- und Bottom-Quarks.
Die Leptonen der ersten Generation sind das Elektron und das Elektron-Neutrino. Die zweite Generation machen das Myon und Myon-Neutrino aus, die dritte das tau und das Tau-Neutrino. Während die Elektronen geladen sind, sind Neutrinos elektrisch neutral.
Die Teilchen verschiedener Generationen ähneln sich in ihren Eigenschaften, sie unterscheiden sich aber in ihrer Masse voneinander: Die Materieteilchen der zweiten und dritten Generation sind schwerer als die der ersten Familie. Zudem sind die Materieteilchen der zweiten und dritten Generation instabil, das heißt sie zerfallen in Teilchen der ersten Familie. Die Materieteilchen der zweiten und dritten Generation, die es in der Frühphase unseres Universums in großen Mengen gab, sind im Laufe der Ausdehnung des Universums in ihre leichteren Geschwister zerfallen.
Jede Form der Materie, aus denen unser Universum besteht, hat eine Form der Antimaterie. Die Materie-Antimaterie-Teilchen hatten sich im frühen Universum gegenseitig vernichtet, bei dem jedoch ein Überschuss an Materie-Teilchen übrigblieb, aus dem unser Universum besteht. Heute besteht die uns umgebende sichtbare Materie ausschließlich aus Teilchen der ersten Generation.
Die Quarks der ersten Generation bilden die Protonen und Neutronen im Atomkern, das Elektron verbleibt als Lepton der ersten Generation in der Atomhülle.
Neben den Materieteilchen gibt es auch die Austauschteilchen, sogenannte Bosonen, die die Wechselwirkungen der Materieteilchen hervorrufen. Die Wechselwirkungen, die zwischen Materieteilchen herrschen, sind die elektromagnetische, die schwache und die starke Kraft.
Auch bei Elementarteilchen kommt eine Chiralität vor, die jedoch nicht mit der Chiralität der Moleküle, wie wir sie beschrieben haben, verwechselt werden darf. Die Chiralität physikalischer Größen lässt sich im Gegensatz zur Chiralität in der Chemie nicht durch eine Spiegelung veranschaulichen. Stattdessen beschreibt sie eine intrinsische, grundlegende Eigenschaft des Teilchens. Sie hängt nicht von Bezugspunkten oder der Perspektive ab (Abb. 18). Die Chiralität ist auch zu unterscheiden vom Spin, also der Bewegungsrichtung des Elementarteilchens. Ich möchte betonen, dass Chiralität eine quantenmechanische Eigenschaft von Elementarteilchen ist. Sie ist eine Eigenschaft der quantenmechanischen Wellenfunktion.
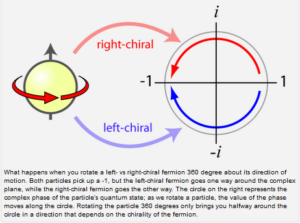
Abb. 18: Ein Teilchen mit Masse hat neben seinem Spin eine bestimmte Chiralität. Ein links-chirales Teilchen kann entweder einen links- oder rechts-Spin haben, je nachdem, in welchem Bezugssystem man sich zum Teilchen befindet. In allen Bezugssystemen ist das Teilchen immer links-chiral, egal welchen Spin es hat. Bei der Chrailität der Masseteilchen handelt es sich um eine quantenmechanische Eigenschaft, bei der ein Teilchen links- oder rechtshändig ist. Konzentrieren wir uns zunächst auf Fermionen, die einen „halben Spin“ haben. Das bedeutet, dass man nicht denselben quantenmechanischen Zustand erhält, wenn man ein Elektron um 360 Grad dreht, sondern denselben Zustand bis zu einem Minuszeichen! Dieses Minuszeichen hängt mit der Quanteninterferenz zusammen. Die Chiralität eines Fermions gibt an, wie es zu diesem Minuszeichen in Form einer komplexen Zahl kommt:
Was passiert, wenn man ein links- vs. rechts-chirales Fermion um 360 Grad um seine Bewegungsrichtung dreht? Beide Teilchen nehmen eine -1 auf, aber das links-chirale Fermion geht in eine Richtung um die komplexe Ebene, während das rechts-chirale Fermion in die andere Richtung geht. Der Kreis auf der rechten Seite stellt die komplexe Phase des Quantenzustands des Teilchens dar; wenn wir ein Teilchen drehen, bewegt sich der Wert der Phase entlang des Kreises. Dreht man das Teilchen um 360 Grad, so bewegt man sich nur auf halbem Weg um den Kreis in einer Richtung, die von der Chiralität des Fermions abhängt. Die Botschaft, die man hier mitnehmen kann, ist, dass Teilchen mit unterschiedlichen Chiralitäten wirklich unterschiedliche Teilchen sind. Wenn wir ein Teilchen mit linkshändigem Spin haben, dann wissen wir, dass es auch eine Version des Teilchens mit rechtshändigem Spin geben sollte. Andererseits muss ein Teilchen mit linkshändiger Chiralität keinen rechtschiralen Partner haben. (Aber es wird auf jeden Fall beide Spins besitzen.)
Die Chiralität eines Elementarteilchens ist entscheidend bei Prozessen der schwachen Kraft, da die vermittelnden W-Bosonen nur an Teilchen mit negativer (linkshändiger) Chiralität und an Antiteilchen mit positiver (rechtshändiger) Chiralität koppeln. Die schwache Kraft wirkt auf alle, auch auf elektrisch ungeladene Elementarteilchen. Sie ist beispielsweise für radioaktive Zerfälle verantwortlich. Die Reichweite der schwachen Kraft ist sehr klein, da die sie vermittelnden Austauschteilchen sehr große Masse haben.
Bei Quarks und den Leptonen Elektron, Myon und Tau gibt es sowohl linkshändige als auch rechtshändige Versionen des Materieteilchens und seines Antimateriepartners. Bislang hat man aber nur linkshändige Neutrinos gefunden. Neutrinos erweisen sich als eine Anomalie, da bisher keine rechtshändigen Neutrinos gefunden wurden (Fermilab 2021, Abb. 19).
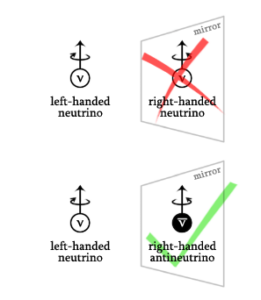
Abb. 19: Bislang hat man aber nur linkshändige Neutrinos gefunden
Daher wurde argumentiert, dass es rechtshändige Neutrinos geben muss, die so genannten „sterilen“ Neutrinos (Dasgupta & Kopp 2021, Abb. 20). Es wurde jedoch die Hypothese aufgestellt, dass die „sterilen“ Neutrinos, wenn sie Massen hätten, die Billionen Mal größer wären als die ihrer normalen linkshändigen Gegenstücke, innerhalb der ersten Sekunden nach dem Urknall oder spätestens bei den Atombildungsprozessen in diese leichteren Neutrinos zerfallen sein müssten. Es blieben also nur noch die linkshändigen Neutrinos übrig. Das könnte die weitere Entwicklung der Händigkeit im Universum und später auch auf den primordialen Protoplaneten beeinflusst haben. Schwere sterile Neutrinos könnten auch die dunkle Materie oder die Dominanz der Materie gegenüber der Antimaterie erklären. Es wurde intensiv nach „sterilen Neutrinos“ gesucht, aber ohne jeden Effekt (Aartsen 2016, Angle 2008, Nature 2015, The LHCb collaboration 2015, Drewes 2013, The STEREO-Collaboration 2023).
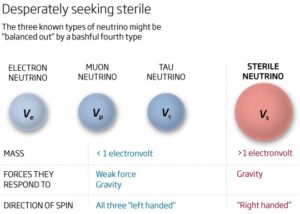
Abb. 20: Sterile Neutrinos
Eine der Theorien, die einen weiteren Beitrag zur Erklärung der Chiralität von Molekülen des Lebens leisten könnte, ist die Theorie der Paritätsverletzung bei schwachen Wechselwirkungen (Lee 1997, Bonner 2000, Wagniere 2007, Bernabeu 2020, Abb. 21).
Paritätsverletzung bezeichnet in der Physik die 1956 entdeckte Tatsache, dass es physikalische Prozesse gibt, die in einer spiegelverkehrt aufgebauten Welt anders ablaufen als in einem Spiegelbild der normalen Welt.
Bis 1956 glaubte man allgemein, dass alle physikalischen Gesetze für das Bild und das Spiegelbild gleich sind, d.h. dass die Parität erhalten bleibt. Die theoretische Vorhersage 1956 (Lee & Yang 1956) der Verletzung der Parität bei schwachen Wechselwirkungen und der experimentelle Beweis dafür einige Monate später (Wu et al. 1957) war nicht nur eine Überraschung für die Forschergemeinde, sondern auch ein Schock.
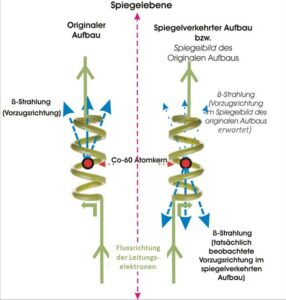
Abb. 21: Prinzip des Nachweises der Paritätsverletzung im Wu-Experiment. Hellblau eingezeichnet sind nur die Elektronen, die in Vorzugsrichtung emittiert werden.
Die ersten experimentellen Arbeiten deuteten darauf hin, dass bei Prozessen im Zusammenhang mit schwachen Wechselwirkungen (z. B. bei der Wechselwirkung von Enantiomeren mit Elektronen, Nukleonen und einigen Nukleonenkomponenten) eine Verletzung der Parität auftritt und Spiegelbilder nicht die gleichen Energien haben. Es wurde festgestellt, dass solche Wechselwirkungen dazu führen, dass eines der Enantiomere etwas stabiler wird als das andere. Dies war eine äußerst wichtige und bahnbrechende Erkenntnis, da sie beispielsweise die asymmetrische Zusammensetzung von Proteinen (die hauptsächlich aus L-Aminosäuren bestehen) und Saccharid-Biopolymeren (die hauptsächlich aus D-Zuckern bestehen) erklären kann.
Später kam man zu dem Schluss, dass alle Atome von Natur aus chiral sind, was genau das Ergebnis von Verletzungen der Kernparität und der chiralen Natur der Elektronen ist (Rubbia 1985, Quack 2002). Es hat sich gezeigt, dass auch schwere Neutrinos und sogar dunkle Materie berücksichtigt werden müssen (Quack 2012, Planckensteriner et al. 2004b). Verschiedene Experimente haben gezeigt, dass selbst Enantiomere von Molekülen nicht völlig energieäquivalent sein können (Quack 2002, 2012, Berger & Quack 2000, Planckensteriner et al. 2005, Abb. 22). Das Universum, wie wir es kennen, besteht aus Materie, nicht aus Antimaterie, und die Verletzung der Parität bei Teilchenzerfällen könnte der Grund dafür sein (Peskin 2002). Andererseits wurde vor kurzem gezeigt (Santos et al. 2020), dass die Energieunterschiede zwischen den Enantiomeren, selbst wenn es eine Paritätsverletzung geben sollte, sehr gering sind, so dass sie schwer zu beobachten sind.
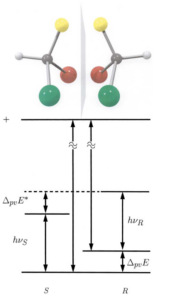
Abb. 22: Die Energien der Enantiomere sind aufgrund einer Symmetrieverletzung unterschiedlich. Die Energiedifferenz ∆pvE0 = ∆pvE und die Reaktionsenthalpie ∆pvH0 = I NA ∆pvE0 I für die Reaktion R = S kann mit dem spektroskopischen Schema Diagramm beschrieben werden. Diese wird auf 10-11 J mol-1 für CHFClBr geschätzt [12]. Wie wichtig ist dieser Energieunterschied Unterschied für die Chemie? Was sind die Konsequenzen für die Biologie? Aus Quack 2002, 2012).
Es gibt aber auch Hinweise, dass der Spin eines Elektrons, also sein Eigendrehimpuls, eine entscheidende Rolle bei der Bestimmung der elektronischen Struktur von Molekülen spielt, weil hier alle Elektronen gepaart sind. Der Spin eines Elektrons kann entweder im Uhrzeigersinn oder gegen den Uhrzeigersinn drehen (Abb. 23). Die Quantenmechanik zeigt, dass zwei Elektronen denselben Platz einnehmen können, wenn beide Elektronen sich in die jeweils andere Richtung drehen. Haben Elektronen denselben Spin, stoßen sie sich gegenseitig ab.
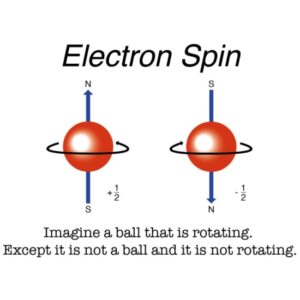
Abb. 23: Spin eines Elektrons
Es ist mittlerweile bekannt, dass der Elektronen-Spin mit der Chiralität der Moleküle zusammenhängt, man bezeichnet es als Chirality-Induced Spin Selectivity (CISS) Effekt. Wandert ein Elektron durch ein chirales Molekül, wird ein Spin bevorzugt. Welcher Spin bevorzugt wird, hängt von der Chiralität des Moleküls ab. Diese Eigenschaft bedeutet, dass bei chiralen Molekülen der Elektronenspin stark an die Molekülform gekoppelt ist. Diese Eigenschaft wird u. a. in der chemischen Industrie genutzt die Enantiomere an magnetischen Oberflächen voneinander zu trennen, um z. B. chemische Erzeugnisse reinzuhalten (Abb. 24).
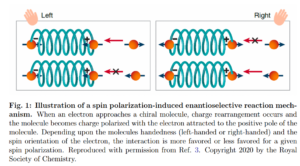
Abb. 24: Illustration eines durch Spinpolarisation induzierten enantioselektiven Reaktionsmechanismus. Wenn sich ein Elektron einem chiralen Molekül nähert, kommt es zu einer Ladungsumlagerung und das Molekül wird ladungspolarisiert, wobei das Elektron vom positiven Pol des Moleküls angezogen wird. Je nach Händigkeit des Moleküls (links- oder rechtshändig) und der Spinausrichtung des Elektrons wird die Wechselwirkung bei einer bestimmten Spinpolarisation mehr oder weniger stark begünstigt.
Es wird angenommen, dass die Bestrahlung magnetisierter Ablagerungen mit UV-Licht unter den präbiotischen Bedingungen der Erde dazu führen könnte, dass spin-polarisierte Elektronen erzeugt wurden, die bestimmte energetisch Enantiomere begünstige. (Dubi 2022, Naaman & Waldeck 2012, Naaman et al. 2020, Ozturk & Sasselov 2022, Abb. 25).
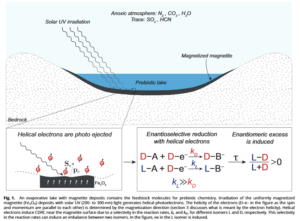
Abb. 25: Ein Verdunstungssee mit Magnetitablagerungen enthält die Ausgangsmoleküle für die präbiotische Chemie. Die Bestrahlung der gleichmäßig magnetisierten Magnetitablagerungen (Fe3O4) mit solarem UV-Licht (200 bis 300 nm) erzeugt spiralförmige Photoelektronen. Die Helizität der Elektronen (D-e- in der Abbildung, da der Spin und der Impuls parallel zueinander sind) wird durch die Magnetisierungsrichtung bestimmt. Helikale Elektronen induzieren CDRC in der Nähe der Magnetitoberfläche aufgrund einer Selektivität der Reaktionsraten kL und kD für verschiedene Isomere Land D. Diese Selektivität in den Reaktionsraten kann ein Ungleichgewicht zwischen zwei Isomeren hervorrufen. In der Abbildung ist ee im L-Isomer induziert.
Die Frage nach dem Ursprung der Chiralität im Leben ist noch offen, hier wird die weitere Forschung zeigen, wann und wie es zur Homochiralität bei Lebewesen kam.
Bemerkenswert ist, dass man von kreationistischer Seite über diese interessanten Erkenntnisse praktisch nichts erfährt (z. B. Junker & Scherer 2006). Stattdessen wird bemerkt, dass global gesehen immer „beide chirale Formen gleichberechtigt“ seien, wobei ein durch physikalisch-chemische Effekte hervorgerufener Symmetriebruch nicht zum Verschwinden einer der beiden Enantiomere aus der Lösung führt (ähnlich Imming 2007a, b). Der Einwand der Kreationisten geht aber an der Sache vorbei, denn – wie gezeigt werden konnte – eine Kompartimentierung erfolgt erwartungsgemäß immer lokal in kleinen, von der Umwelt abgeschotteten Bereichen. Zur Synthese eines homochiralen Kettenmoleküls muss das andere Enantiomer auch nicht vollständig „verschwinden“ – es genügt, wenn sich durch eine mehr oder weniger stark ausgeprägte Asymmetrie die Wahrscheinlichkeit der Synthese homochiraler Moleküle lokal erhöht. Findet sich in einem Molekül-Ensemble ein homochirales Molekül mit einer charakteristischen Funktion (z. B. als Katalysator oder Matrize für die Synthese weiterer homochiraler Spezies), ist das Ergebnis ein Vorgang, der in eine Selektion hineintreibt. Von dem Zeitpunkt an, an dem ein homochirales Kompartiment (wo und aufgrund welcher Eigenschaften auch immer) „das Rennen machte“, waren die Würfel für eine andauernde Diskriminierung des anderen Enantiomers gefallen. Ein selektionäres Szenario ist auch nicht besonders spekulativ, sondern unter den gegebenen Voraussetzungen eine plausible theoretische Erwartung.
An dieser Stelle haben wir die chemischen Grundlagen der Entstehung von Biomolekülen ausreichend besprochen. Eine offene Frage bleibt tatsächlich der Ort der Lebensentstehung. Schon in unserem Beitrag zur Geschichte der Abiogenese-Forschung hatten wir erfahren, dass einige die Entstehung des Lebens im Weltall vermuten, welches unter dem Begriff der Panspermie zusammengefasst wird. Mit dieser wollen wir uns im nächsten Beitrag befassen.
Literatur
Aartsen, M.G. (2016): Searches for Sterile Neutrinos with the IceCube Detector. Phys. Rev. Lett. 117:071801.
Aliashkevich, A., Alvarez, L., Cava, F. (2018): New Insights Into the Mechanisms and Biological Roles of D-Amino Acids in Complex Eco-Systems. Front. Microbiol. 9: 683.
Alvarez, L., Aliashkevich, A., de Pedro, M. A., and Cava, F. (2018). Bacterial secretion of D-arginine controls environmental microbial biodiversity. ISME J. 12:438–450.
Angle, J. (2008): The XENON10 Collaboration. Limits on spin-dependent WIMP-nucleon cross-sections from the XENON10 experiment. Phys. Rev. Lett. 101:091301.
Asakura, K., Kurihara, K, Ikumo, A. et al. (2000): Chirally autocatalytic reaction performed in highly supersaturated conditions. Macromolecular Symposia 160: 7–13.
Avnir, G. (1994): Peptides containing a D-amino acid from frogs and molluscs. J. Biol. Chem. 269:10967–10970.
Bada, J.L. (2009): Enantiomeric excesses in the Murchison meteorite and the origin of homochirality in terrestrial biology. PNAS USA 106, E85.
Bastings, J.J.A.J., van Eik, H.M., Olde Damink, S.W., Rensen, S.S. (2019): D-amino Acids in Health and Disease: A Focus on Cancer. Nutrients 11:2205.
Bailey, J. (2000): Chirality and the origin of life. Acta Astronautica 46, 627–631.
Berger, R., Quack, M. (2000): Electroweak quantum chemistry of alanine: Parity violation in gas and condensed phases. Chem. Phys. Chem. 1:57–60.
Bernabeu, J. (2020): Symmetries and Their Breaking in the Fundamental Laws of Physics. Symmetry 12:1316.
Blackmond, D. G., Klussmann, M. (2007): Spoilt for choice: assessing phase behavior models for the evolution of homochirality. Chemical Communications, (39), 3990.
Bonner, W. (2000): Parity violation and the evolution of biomolecular homochirality. Chirality 12:114–126.
Bonner, W.A., Bean, B.D. (2000): Asymmetric photolysis with elliptically polarized light. Orig Life Evol Biosph. 30(6):513-7.
Breslow, R., Cheng, Z.L. (2010): L-amino acids catalyze the formation of an excess of D-glyceraldehyde, and thus of other D sugars, under credible prebiotic conditions. PNAS USA. 107(13):5723-5.
Brückner, H., Fujii, N. (Eds. 2011): D-Amino Acids in Chemistry, Life Sciences, and Biotechnology; Wiley-VCH, Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2011
Cava, F., de Pedro, M. A., Lam, H., Davis, B. M., and Waldor, M. K. (2011): Distinct pathways for modification of the bacterial cell wall by non-canonical D-amino acids. EMBO J. 30:3442–3453.
Chen, Y., Ma, W. (2020): The origin of biological homochirality along with the origin of life. PLoS Comput Biol 16(1): e1007592.
Corrigan, J.J. (1969): D-Amino Acids in Animals. Science 164:142–149.
Dasgupta, B., Kopp, J. (2021): Sterile neutrinos. Phys.Rep. 928: 1–63.
Deamer, D. (2017): The role of lipid membranes in life´s origin. Life 7:5.
Deamer, D., Dick, R., Thiemann, W., Shinitzky, M. (2007): Intrinsic Asymmetries of Amino Acid Enantiomers and Their Peptides: A Possible Role in the Origin of Biochirality. Chirality 19:751–763.
Devínsky, F. (2021): Chirality and the Origin of Life. Symmetry 13:2277.
Drewes, M. (2013): The Phenomenology of Right Handed Neutrinos. Int. J. Mod. Phys. E 22:1330019.
Dubi, Y. (2022): Spinterface chirality-induced spin selectivity effect in bio-molecules. Chem. Sci. 13:10878
Fajszi, C., Czege, J. (1981): Critical evaluation of mathematical models for the amplification of chirality. Origins of Life and Evolution of the Biosphere 11:143–162.
Fermilab (2021): All Things Neutrino. Fermilab. Available online: https://neutrinos.fnal.gov/
Fisher, G.H., Petrucelli, L., Gardner, C., Emory, C., Frey, W.H., Amaducci, L., Sorbi, S., Sorrentino, G., Borghi, M., D’Aniello, A. (1994): Free D-amino acids in human cerebrospinal fluid of Alzheimer disease, multiple sclerosis, and healthy control subjects. Mol. Chem. Neuropathol. 23:115–124.
Fisher, G.H., Torres, D., Bruna, J., Cerwinski, S., Martin, T., Bergljung, C., Gruneiro, A., Chou, S.J., Man, E.H., Pappatheodorou, S. (1995): Presence of D-aspartate and D-glutamate in tumor protein. Cancer Biochem. Biophys. 15:79–82.
Fletcher, S. P. , Jagt, R.B.C., Feringa, B. (2007): An astrophysically-relevant mechanism for amino acid enantiomer enrichment. Chem. Commun., 2007, 2578 —2580
Follmann, H. (1981): Chemie und Biochemie der Evolution. Wie und Wo entstand das Leben? Heidelberg.
Foltyn, V.N., Bendikov, I., De Miranda, J., Panizzutti, R., Dumin, E., Shleper, M., Li, P., Toney, M.D., Kartvelishvily, E., Wolosker, H. (2005): Serine racemase modulates intracellular D-serine levels through an alpha,beta-elimination activity. J. Biol. Chem. 280:1754–1763.
Friedman, M. (2010): Origin, microbiology, nutrition, and pharmacology of D-amino acids. Chem. Biodivers. 7:1491–1530.
Fujii, N. (2002): D-amino acids in living higher organisms. Orig. Life Evol. Biosph. 32:103–127.
Fujii, N., Takata, T., Fujii, N., Aki, K., Sakaue, H. (2018): D-amino acids in protein: The mirror of life as a molecular index of aging. BBA-Proteins Proteom. 1866:840–847.
Gaspar, D., Veiga, A.S., Castanho, M.A.R.B. (2013): From antimicrobial to anticancer peptides. A review. Front. Microbiol. 4:294.
Genchi, G. (2017): An overview on D-amino acids. Amino Acids 49:1521–1533.
Graham, D.L., Beio, M.L., Nelson, D. L., Berkowitz, D.B. (2019): Human Serine Racemase: Key Residues/Active Site Motifs and Their Relation to Enzyme Function. Front. Mol. Biosci. 6:8.
Grishin, D.V., Zhdanov, D.D., Pokrovskaya, M.V., Sokolov, N.N. (2020): D-amino acids in nature, agriculture and biomedicine. Front. Life Sci. 13:11–22.
Gusev, V.A. (2001): Living Universe. Fundamental of Life; Chap-IV-06; Elsevier: Paris.
Gutiérrez, M., Capson, T., Guzmán, H.M., Quiňoá, E., Riguera, R. (2004): L-Galactose as a natural product: Isolation from a marine octocoral of the first α-L-galactosyl saponin. Tetrahedron Lett. 45:7833–7836.
Harada, K. (1970): Origin and development of optical activity of organic compounds on the primordial earth. Naturwissenschaften 57, 114–119.
Hashimoto, A., Nishikawa, T., Oka, T., Takahashi, K. (1993a): Endogenous D-serine in rat brain: N-methyl-D-aspartate receptor-related distribution and aging. J. Neurochem. 60:783–786.
Hashimoto, A., Nishikawa, T., Konno, R., Niwa, A., Yasumura, Y., Oka, T., Takahashi, K. (1993b): Free D-serine, D-aspartate and D-alanine in central nervous system and serum in mutant mice lacking D-amino acid oxidase. Neurosci. Lett. 152:33–36.
Hashimoto, A., Oka, T. (1997): Free D-aspartate and D-serine in the mammalian brain and periphery. Prog. Neurobiol. 52:325–353.
Hazen, R.M. (2001): Life’s rocky start. Scientific American 284, 63–71.
Hazen, R.M., Filley, T.R., Goodfriend, G.A. (2001): Selective adsorption of L- and D-amino acids on calcite: implication for biochemical homochirality. PNAS 98:5487–5490.
Hein, J.E., Tse, E., Blackmond, D.G. (2011): A route to enantiopure RNA precursors from nearly racemic starting materials. Nat Chem. 3(9):704-6.
Hillig, W. B., Turnbull, D. (1956): Theory of crystal growth in undercooled pure liquids. Journal of Chemical Physics 24, 914.
Hills, G. M. (1949): Chemical factors in the germination of spore-bearing aerobes; the effects of amino acids on the germination of Bacillus anthracis, with some observations on the relation of optical form to biological activity. Biochem. J. 45:363–370.
Hobbs, P. V., Ketcham, W. M. (1969): The planar growth of ice from pure melt. Physics of ice. Proceedings of the International Symposium on Physics of Ice, München, 95–112.
Hochbaum, A. I., Kolodkin-Gal, I., Foulston, L., Kolter, R., Aizenberg, J., Losick, R. (2011): Inhibitory effects of D-amino acids on Staphylococcus aureus biofilm development. J. Bacteriol. 193:5616–5622.
Ikehara, K. (2005): Possible steps to the emergence of life: The [GADV]-protein world hypothesis. Chem. Rec. 5:107–118.
Imming, P. (2007a): Die fehlenden Spiegelbilder. www.genesisnet.info/index.php?Artikel=42081&Sprache=de&l=1.
Imming, P. (2007b): Artikel über Chiralität: Wurde wichtige neuere Literatur unterschlagen? www.genesisnet.info/index.php?News=80.
Jin, D., Miyahara, T., Oe, T., Toyo’oka, T. (1999): Determination of D-amino acids labeled with fluorescent chiral reagents, R(-)- and S(+)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylami nosulfonyl)-2,1,3-benzoxadiazoles, in biological and food samples by liquid chromatography. Anal. Biochem. 269:124–132.
Jenkins , J.K., Salam, A., Thirunamachandran, T. (1994): Discriminatory dispersion interactions between chiral molecules. Molecular Physics 82:835–840.
Jung, G. (1992): Proteine aus der D-chiralen Welt. Angew. Chem. 104:1484–1486.
Junker, R., Scherer, S. (2006): Evolution. Ein kritisches Lehrbuch. Gießen.
Kimura, H. (2000): Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267, 129–133.
Kimura, Y., Mikami, Y., Osumi, K., Tsugane, M., Oka, J., Kimura, H. (2013): Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. 27, 2451–2457.
Kirkpatrick, R.J. (1975): Crystal growth from melt: a review. American Mineralogist 60:798–814.
Konno, R., Brückner, H., D’Aniello, A., Fischer, G., Fujii, N., Homma, H. (Eds. (2007): D-Amino Acids: A New Frontier in Amino Acids and Protein Research, Practical Methods and Protocols; Nova Science Publishers, Inc.: New York, NY, USA
Kreil, G. (1994): Conversion of L- to D-Amino Acids: A Posttranslational Reaction. Science 266:996–997.
Lam, H., Oh, D.-C., Cava, F., Takacs, C. N., Clardy, J., de Pedro, M. A., et al. (2009): D-Amino acids govern stationary phase cell wall remodeling in bacteria. Science 325:1552–1555.
Lee, T.D. (1997): Obituary: Chien-Shiung Wu (1912-97), Experimental physicist, co-discoverer of parity violation. Nature 386:334.
Lee, T.D., Yang, C.N. (1956): Question of parity conservation in weak interactions. Phys. Rev. 104:254–258.
Masters, P.M., Bada, J.L., Zigler, J.S., Jr. (1977): Aspartic acid racemisation in the human lens during ageing and in cataract formation. Nature 268:71–73.
Masters, P.M., Bada, J.L., Zigler, J.S., Jr. (1978): Aspartic acid racemization in heavy molecular weight crystallins and water insoluble protein from normal human lenses and cataracts. Proc. Natl. Acad. Sci. USA 75:1204–1208.
Mauksch, M., Shengwei, W., Freund, M., Zamfir, A., Tsogoeva, S.B. (2010): Spontaneous Mirror Symmetry Breaking in the Aldol Reaction and its Potential Relevance in Prebiotic Chemistry. Orig. Life Evol. Biosph. 40:79–91.
Mor, A., Amiche, M., Nicolas, P. (1992): Enter a new posttranslational modification: D-amino acids in gene-encoded peptides. Trends Biochem. Sci. 17:481–485.
Morowitz, H. (1969): Mechanism for the amplification of fluctuations in racemic mixtures. J. Theor. Biol. 25:491–494.
Mortimer, C. E., Müller, U. (2019): Chemie. 13. Auflage. Stuttgart: Thieme Verlag
Mustafa, A.K., Kim, P.M., Snyder, S.H. (2004): D-Serine as a putative glial neurotransmitter. Neuron Glia Biol. 1:275–281.
Mustafa, A. K., Ahmad, A. S., Zeynalov, E., Gazi, S. K., Sikka, G., Ehmsen, J. T., et al. (2010): Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J. Neurosci. 30:1413–1416.
Mutaguchi, Y., Ohmori, T., Akano, H., Doi, K., Ohshima, T. (2013): Distribution of D-amino acids in vinegars and involvement of lactic acid bacteria in the production of d-amino acids. SpringerPlus 2:691.
Naaman, R., Paltiel, Y., Waldeck, D. H. (2020): Chiral Induced Spin Selectivity Gives a New Twist on Spin-Control in Chemistry. Acc. Chem. Res. 53:2659−2667
Naaman, R., Waldeck, D. H. (2012): Chiral-Induced Spin Selectivity Effect. The Journal of Physical Chemistry Letters, 3(16), 2178–2187.
Nagata, Y., Borghi, M., Fisher, G.H. (1995): Free D-serine concentration in normal and Alzheimer human brain. Brains Res. Bull. 38:181–183.
Nanita, S.C., Cooks, R.G. (2006): Serine octamers: Clusters formation. Reaction, and implication for biomolecule homochirality. Angew. Chem. Int. Ed. Engl. 45:554–569.
Nature (2015): Only left-handed particles decay. Nature 524:8.
Ohide, H., Miyoshi, Y., Maruyama, R., Hamase, K., Konno, R. (2011): D-Amino acid metabolism in mammals: Biosynthesis, degradation and analytical aspects of the metabolic study. J. Chrom. B Analyt. Technol. Biomed. Life Sci. 879:3162–3168.
Ozturk, S. F., Sasselov, D. D. (2022): On the origins of life’s homochirality: Inducing enantiomeric excess with spin-polarized electrons. PNAS USA 19(28): e2204765119
Peskin, R. (2002): The matter with antimatter. Nature 419:25–27.
Pikuta, E.V., Hoover, R.B., Klyce, B., Davies, P.-C.W., Davies, P. (2006): Bacterial utilization of L-sugars and D-amino acids. Proceedings 6309:63090A.
Pikuta, E. V., Menes, R. J., Bruce, A. M., Lyu, Z., Patel, N. B., Liu, Y., et al. (2016): Raineyella antarctica gen. nov., sp. nov., a psychrotolerant, D-aminoacid-utilizing anaerobe isolated from two geographic locations of the Southern Hemisphere. Int. J. Syst. Evol. Microbiol. 66:5529–5536.
Pizzarello, S., Weber, A.L. (2004): Prebiotic amino acids as asymmetric catalysts. Science 303:1151.
Plankensteiner, K., Reiner, H., Rode, B.M. (2004): From earth’s primitive atmosphere to chiral peptides–the origin of precursors for life. Chem Biodivers. 1(9):1308-15.
Plankensteiner, K., Righi, A., Rode, B.M., Gargallo, R., Jaumot, J., Tauler, R. (2004b): Indications toward a stereoselectivity of the salt-induced peptide formation reaction. Inorg. Chim. Acta 357:649–656.
Plankensteiner, K., Reiner, H., Rode, B.M. (2005): Stereoselective differentiation in the salt-induced formation reaction and its relevance for the origin of life. Peptides 26:535–541.
Plasson, R., Kondepudi, D.K., Bersini, H., Commeyras, A., Asakura, K. (2007): Emergence of homochirality in far-from-equilibrium systems: Mechanisms and role in prebiotic chemistry. Chirality 19:589–600.
Podlech, J. (2001): Origin of organic molecules and biomolecular homochirality. Cell. Moll. Life Sci. 58:44–60.
Pundir, C.S., Lata, S., Narval, V. (2018): Biosensors for determination of D- and L-amino acids: A Review. Biosens. Bioelectron. 117:373–384.
Quack, M. (2002): How important is parity violation for molecular and biomolecular chirality? Angew. Chem. Int. Ed. Engl. 41:4618–4630.
Quack, M. (2012): Molecular Parity Violation and Chirality: The Asymmetry of Life and the Symmetry Violation in Physics. In Quantum Systems in Chemistry and Physics: Progress in Methods and Applications; Chapter 3; Theoretical Chemistry and Physics; Nishikawa, K., Maruani, J., Brändas, E.J., Delgado-Barrio, G., Piecuch, P., Eds.; Springer Science: Dordrecht, The Netherlands, Volume 26, pp. 47–76.
Raboni, S., Marchetti, M., Faggiano, S., Campanini, B., Bruno, S., Marchesani, F., Margiotta, M., Mozzarelli, A. (2019): The Energy Landscape of Human Serine Racemase. Front. Mol. Biosci. 5:112.
Ramon-Perez, M. L., Diaz-Cedillo, F., Ibarra, J. A., Torales-Cardena, A., Rodriguez-Martinez, S., Jan-Roblero, J., et al. (2014): D-Amino acids inhibit biofilm formation in Staphylococcus epidermidis strains from ocular infections. J. Med. Microbiol. 63:1369–1376.
Rein, D. (1993): Die wunderbare Händigkeit der Moleküle. Basel.
Reiner, R., Plankensteiner, K., Fitz, D., Rode, B. M. (2006): The possible influence of L-histidine on the origin of the first peptides on the primordial earth. Chemistry & Biodiversity 3:611–621.
Riehl, J.P. (2010): Mirror-Image Asymmetry: An Introduction to the Origin and Consequences of Chirality; J. Wiley&Sons: Hoboken, NJ, USA.
Robinson, T. (1976): D-amino acids in higher plants. Life Sci. 19:1097–1102.
Rohde, H., Burdelski, C., Bartscht, K., Hussain, M., Buck, F., Horstkotte, M. A., et al. (2005): Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol. 55:1883–1895.
Rubbia, C. (1985): Experimental observation of the intermediate vector bosons W+,W− and Z0. Rev. Mod. Phys. 57:699–722.
Rumbo, C., Vallejo, J. A., Cabral, M. P.,Martínez-Guitián, M., Pérez, A., Beceiro, A., et al. (2016): Assessment of antivirulence activity of several d-amino acids against Acinetobacter baumannii and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 71:1–9.
Santos, A.C.L., Muniz, C.R., Oliveira, L.T., Souza, J.T. (2020): Contribution of a modified electrodynamics to the molecular biochirality. Chirality 32, 1186–1190.
Sasabe, J., Miyoshi, Y., Rakoff-Nahoum, S., Zhang, T., Mita, M., Davis, B.M., Hamase, K., Waldor, M.K. (2016): Interplay between microbial D-amino acids and host D-amino acid oxidase modifies murine mucosal defence and gut microbiota. Nat. Microbiol. 1:16125.
Schalley, C. A., Weis, P. (2002): Unusually stable magic number clusters of serine with a surprising preference for homochirality. International Journal of Mass Spectrometry 221:9–19.
Schell, M.J., Cooper, O.B., Snyder, S.H. (1997): D-aspartate localizations imply neuronal and neuroendocrine roles. Proc. Natl. Acad. Sci. USA 94:2013–2018.
Schell, M.J., Molliver, M.E., Snyder, S.H. (1995): D-Serine, an endogenous synaptic modulator leocalization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. USA 92:3948–3952.
Sela, M., Zisman, E. (1997): Different roles of D-amino acids in immune phenomena. FASEB J. 11:449–456.
Shinitzky, M., Nudelman, F., Carda, Y. et al. (2002): Unexpected differences between D- and L-Tyrosine lead to chiral enhancement in racemic mixtures. Origins of Life and Evolution of the Biosphere 32: 285–297
Soai, K., Sato, I., Shibata, T. (2001) Asymmetric autocatalysisand the origin of chiral homogeneity in organic compounds. Chem Rec 1:321–332.
Soai, K., Shibata, T., Morioka, H., Choji, K. (1995) Asym-metric autocatalysis and amplification of enantiomeric excessof a chiral molecule. Nature 378:767–768.
Stevens, C.M., Halpern, P.E., Gigger, R.P. (1951): Occurrence of d-amino acids in some natural materials. J. Biol. Chem. 190:705–710.
Suzuki, N., Itabashi, Y. (2019): Possible roles of amphiphilic molecules in the origin of biological homochirality. Symmetry 11:966.
Tarasevych, A.V., Sorochinsky, A.E, Kukhar, V.P, Guillemin, J.C. (2013): Deracemization of amino acids by partial sublimation and via homochiral self-organization. Orig Life Evol Biosph. 43(2):129-35.
The LHCb collaboration, Aaij, R., Raven, G. (2015): Determination of the quark coupling strength |Vub| using baryonic decays. Nat. Phys. 11:743–747.
The STEREO Collaboration (2023): STEREO neutrino spectrum of 235U fission rejects sterile neutrino hypothesis. Nature 613:257–261.
Trinks, H. (2001): Auf den Spuren des Lebens. Bericht zur Expedition in das Eis von Spitzbergen vom 17. Mai 1999 bis 14. September 2000. Aachen.
Uda, K., Abe, K., Dehara, Y., Mizobata, K., Sogawa, N., Akagi, Y., Saigan, M., Radkov, A.D., Moe, L.A. (2016): Distribution and evolution of the serine/aspartate racemase family in invertebrates. Amino Acids 48:387–402.
Viedma, C. (2001): Enantiomeric Crystallization from DL-Aspartic and DL-Glutamic Acids: Implications for Biomolecular Chirality in the Origin of Life. Orig Life Evol Biosph 31:501–509.
Wagniére, G.H. (2007): On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image; Wiley-VCH: Zürich, Switzerland
Weber, A.L., Pizzarello, S. (2006): The peptide-catalyzed stereospecific synthesis of tetroses: A possible model for prebiotic molecular evolution. Proc. Natl. Acad. Sci. USA 103:12713–12717.
Wolosker, H., Blackshaw, S., Snyder, S.H. (1999): Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 96:13409.
Wu, C.S., Ambler, E., Hayward, R.W., Hoppes, D.D., Hudson, R.P. (1957): Experimental Test of Parity Conservation in Beta Decay. Phys. Rev. 105:1413–1415.
Wu, M., Walker, S. I., & Higgs, P. G. (2012): Autocatalytic Replication and Homochirality in Biopolymers: Is Homochirality a Requirement of Life or a Result of It? Astrobiology, 12(9), 818–829.
Yu, C., Li, X., Zhang, N., Wen, D., Liu, C., Li, Q. (2016): Inhibition of biofilm formation by d-tyrosine: effect of bacterial type and d-tyrosine concentration. Water Res. 92:173–179.
https://internet-evoluzzer.de/standardmodell-der-teilchenphysik/
https://www.weltmaschine.de/physik/standardmodell_der_teilchenphysik/
[1] Vgl auch: https://internet-evoluzzer.de/standardmodell-der-teilchenphysik/