UPDATE 16.04. 2020
Der Artikel wurde überarbeitet und in mehrere Videos geteilt. Siehe hier:
Youtube-Video: Phylogenetische Systematik Teil 1: Taxonomie
Youtube-Video: Phylogenetische Systematik Teil 2: Kladistik
Youtube-Video: Phylogenetische Systematik Teil 3: Stammbaum des Lebens
Phylogenetische Systematik Teil 4: Homologie
Befasst man sich mit Evolution, kommt man an einem bestimmten Thema nicht vorbei: der phylogenetischen Systematik. Jeder hat sicherlich den ein oder anderen evolutionären Stammbaum gesehen. Unten stehend ist ein Stammbaum der Primaten dargestellt (Abb. 1). Solche Stammbäume stellen die Verwandtschaftsbeziehungen zwischen den untersuchten Gruppen der Lebewesen dar; zeigen also, welche Arten auf einen gemeinsamen Vorfahren zurückgehen. Das Ziel dieses Artikels ist es darzustellen nach welchen Regeln und Prinzipien man solche evolutionären Stammbäume erstellt.
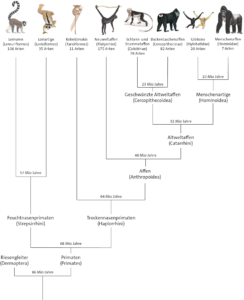
Abb. 1: Stammbaum der Primaten. Quelle: Deutsches Primatenzentrum
1. Grundlagen der Systematik und Taxonomie
Bevor wir jedoch mit den erstellen von Stammbäumen beginnen können, müssen wir ganz klein anfangen. Bevor man nämlich Stammbäume erstellen kann, muss man eigentlich wissen von welchen Tieren oder Pflanzen man einen Stammbaum erstellen will. Die Zahl der unterschiedlichen Lebensformen ist mannigfaltig und diese muss erst einmal geordnet werden. Hier sind zwei Begriffe von Wichtigkeit: Systematik und Taxonomie. Den meisten Menschen sind die Kriterien wie man Lebewesen einordnet und klassifiziert nicht bekannt. Dieses Wissenschaftsfeld, also die Benennung und Klassifizierung eines Lebewesens, bezeichnet man als Taxonomie. Die Systematik hingegen ist die Wissenschaft der Vielfalt der Organismen, die neben der Taxonomie auch die Phylogenie (evolutionäre Verwandtschaft) und die Biogeographie (wo leben die einzelnen Arten) miteinschließt.
Das mag den meisten Nicht-Biologen vielleicht wie ein Fremdwort klingen, doch wird der ein oder andere sicherlich mal aufgeschnappt haben, dass die wissenschaftlichen Namen bestimmter Arten aus zwei meist lateinischen oder griechischen Wörtern bestehen, die oftmals kursiv geschrieben werden:
Panthera leo ist der wissenschaftliche Artname für den Löwen, Canis lupus für den Wolf und Homo sapiens für uns Menschen. „Löwe“, „Wolf“ und „Mensch“ werden in der Taxonomie als Trivialnamen bezeichnet, da sie in anderen Sprachen andere Bezeichnungen haben (z. B. im Englischen: „lion“, „wolf“, „human“). Die wissenschaftliche Bezeichnung wird jedoch überall anerkannt. Das ist der Vorteil dieser Form der Nomenklatur. Interessanterweise sind für uns bei einer Reihe ausgestorbener Arten die wissenschaftlichen Namen geläufiger als die deutschen oder englischen; namentlich sind hier die Dinosaurier zu erwähnen: Jeder kennt Tyrannosaurus rex, seine deutsche Bezeichnung – König der Tyrannenechsen – findet praktisch keine Erwähnung. Häufig liest man auch die Schreibweise T. rex oder H. sapiens, also bei dem der erste Name abgekürzt wird. (Achtung absolutes Nerd-Wissen: Die Schreibweise T-rex, also mit Bindestrich, ist falsch).
Diese binäre Nomenklatur lässt sich auf den schwedischen Naturforscher Carl von Linné zurückführen, der 1735 sein Werk mit dem Titel „Systema Naturae“ publizierte. Bis 1768 erfuhr diese Arbeit ihre 12. Auflage und Linné beschrieb über 7.700 Pflanzen- und 6.200 Tierarten.
Das Prinzip der binären Nomenklatur ist streng hierarchisch. Die kleinste Einheit bildet die Art. Ähnliche Arten werden in Gattungen, ähnliche Gattungen in Familien, ähnliche Familien in Klassen und ähnliche Klassen in Stämme eingeteilt. Folgendes Beispiel anhand des Löwen und des Gänseblümchens sollen dies verdeutlichen:
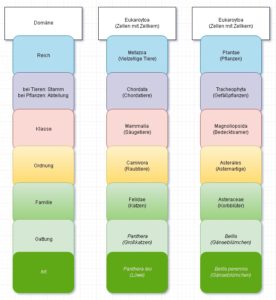
Abb.2: Systematische Einteilung beim Löwen und Gänseblümchen.
Aber nur die Art stellt ein natürliches System dar: sie sind biologische Realitäten. Alle Ränge oberhalb der Art werden als künstliches System angesehen, die ähnliche Gruppen miteinander verbinden. Neben dieser in Abb. 2 dargestellten Ränge gibt es noch Untergruppen: Klassen können z. B. in Unterklassen eingeteilt werden, Gattungen in Gattungsgruppen (sog. Tribus) oder Arten in Unterarten. Vom Löwen werden z. B. verschiedene Unterarten anerkannt; der Indische Löwe heißt dann z. B. Panthera leo persica, der eiszeitliche ausgestorbene Höhlenlöwe hat den Namen Panthera leo spelaea, der ostafrikanische Massai-Löwe Panthera leo massaicus. Es handelt sich hierbei um ein und dieselbe Art, die jedoch in geographische Populationen eingeteilt wird. Zur Problematik, was überhaupt eine Art ist, wird in einem anderen Artikel näher untersucht.
Innerhalb dieser binären Nomenklatur kann, wenn neue Erkenntnisse hinzukommen, eine Gattung in mehrere Gattungen aufgespalten werden bzw. mehrere Gattungen in eine zusammengefasst werden, Unterarten können als Arten anerkannt werden etc. Dies läuft jedoch nicht willkürlich ab, sondern folgt festen Regeln. Ob z. B. eine Art in eine andere Gattung gestellt wird, hängt natürlich von den untersuchten Merkmalen ab.
Um die Arten richtig zu klassifizieren gibt es feste Regeln, von denen man nicht abweichen soll. So gibt es z. B. die Internationalen Regeln für die Zoologische Nomenklatur (International Code of Zoological Nomenclature, ICZN). Hierbei handelt es sich um eine Konvention, durch die die Benennung und Klassifikation aller Tierarten international geregelt wird. Dieser kann hier eingesehen werden. Mit über 90 Artikeln in 18 Kapiteln, bei dem die Artikel auch in mehrere Unterpunkte gegliedert sind, wirkt es sehr kompliziert und hat dem ein oder anderen Taxonomen sicherlich Kopfschmerzen bereitet. Für die Botanik gibt es ein entsprechendes Gegenstück.
So legt der Artikel 5 fest, dass der Artname aus zwei Namen bestehen soll: Dem Gattungsnamen (z. B. Panthera) und dem Artnamen (sog. Epitheton). Der Anfangsbuchstabe des Gattungsnamen wird großgeschrieben, das Epitheton klein. Hat man eine Unterart wird ein dritter Name (ebenfalls kleingeschrieben) eingefügt (z. B. Panthera leo massaicus).
Gibt es in einer Gattung mehrere Arten und diese werden aufgezählt, reicht es den Gattungsnamen abzukürzen: z. B. Panthera leo kann als P. leo abgekürzt werden, Tyrannosaurus rex als T. rex (womit die Abkürzung T-rex, also mit Bindestrich, den internationalen Regeln nicht entspricht).
Wird von irgendeinem Rang gesprochen, benutzt man häufig das Wort Taxon (plural: Taxa): Eine Gattung ist ebenso ein Taxon, wie eine Familie etc.
Das mag dem Nicht-Biologen müßig erscheinen und sicherlich wirkt es schon fast bürokratisch. Aber bei aller Kritik ist es heute die effizienteste Methode in das Chaos der biologischen Vielfalt eine gewisse Ordnung zu bringen.
Linné war jedoch kein Anhänger der Evolution, sondern glaubte an die Konstanz der Arten. Linné ordnete die verschiedenen Tiere und Pflanzen nach ihren Merkmalen, evolutionäre Ursprünge kamen ihm dabei nicht in den Sinn. Umso verblüffender ist es festzustellen, dass Linné unsere Art, H. sapiens, zur Ordnung der Primaten zählte, zu der die Affen gehören. Wenn also beliebiger Kreationist das Kinderlied „I’m No Kin To The Monkey“ singt, dem soll gesagt werden, dass die Schuld nicht (nur) bei Darwin zu suchen ist, sondern auch beim Kreationisten Linné.
Dass Evolutionsverlauf und Verwandtschaftsbeziehungen früher keine große Rolle spielten, zeigt sich z. B. darin, dass Elefanten, Flusspferde und Nashörner als Dickhäuter (Pachydermes) zusammengefasst wurden, obwohl wir heute wissen, dass alle drei Gruppen unterschiedlichen evolutionären Linien entsprangen. Elefanten sind näher mit den Schliefern und Seekühen verwandt, Nashörner werden mit den Pferden und Tapiren zu den Unpaarhufern zusammengefasst und Flusspferde sind Paarhufer, deren nächsten heute lebenden Verwandten die Wale sind.
Es braucht also ein Ordnungsprinzip, dass den Evolutionsverlauf und die Verwandtschaftsbeziehungen berücksichtigt, denn nicht Merkmale definieren Gruppen, sondern die Abstammung. Es muss also untersucht werden, welche Merkmale geeignet sind die Verwandtschaftsbeziehungen zu ermitteln. Hier bedient man sich der Methoden der phylogenetischen Systematik.
2. Phylogenetische Systematik
In den 1960er Jahren hatte der deutsche Morphologe Willi Hennig ein System ausgearbeitet, welches die verwandtschaftlichen Beziehungen zwischen den Organismen anhand von Merkmalsvergleichen ermitteln konnte. Mein bezeichnet sich allgemein als Kladistik oder Phylogenetische Systematik (HENNIG 1966). Ein jeder Organismus, will man ihn beschreiben, hat für ihn typische charakteristische Eigenschaften, die man als Merkmale definiert. Diese können Strukturmerkmale sein (also die Anatomie), aber auch physiologische (z. B. Warmblütigkeit), ökologische (wasser- oder landlebend) oder ethologische (Verhaltensweisen). Durch einen Vergleich diverser Merkmale lassen sich die Verwandtschaftsgrade der Organismen ableiten. Gehen die Merkmale auf einen gemeinsamen evolutionären Ursprung zurück, spricht man von Homologien.
Der Biologe Adolf Remane hatte in den 1950er Jahren die Homologie-Kriterien festgelegt, die sich auch heute noch in den Biologie-Lehrbüchern wiederfinden und von weiteren Autoren verfeinert und ergänzt wurden (REMANE 1952).
Im wesentlichen sind drei Homologie-Kriterien bekannt:
Kriterium der Lage
Morphologische (oder andere) Strukturen sind dann zueinander homolog, wenn sie trotz unterschiedlicher Ausprägung in Gestalt und Anzahl in einem vergleichbaren Gefügesystem stets die gleiche Lagebeziehung aufweisen.
Ein bekanntes Beispiel hierfür sind die Vordergliedmaßen der Wirbeltiere (siehe Abb.3). Egal ob wir einen Menschen, ein Pferd, einen Vogel oder eine Echse haben, so ist der Grundaufbau der Vordergliedmaßen vom Prinzip her gleich: ein Oberarmknochen, zwei Unterarmknochen, mehrere Handwurzelknochen, bis zu fünf Mittelhandknochen und bis zu fünf Finger. Selbiges gilt entsprechend für die Hintergliedmaßen. Trotz des gleichen Grundaufbaus kam es im Verlauf der Erdgeschichte zu verschiedenen Umwandlungen, da die Gliedmaßen unterschiedliche Zwecke erfüllen: laufen, graben, schwimmen, fliegen etc. Einzelne Knochen verschmolzen oder wurden reduziert, die Proportionen änderten sich, doch der Grundaufbau bleibt der gleiche und dieser weist auf gemeinsame Vorfahren hin, von denen aus sich die Entwicklung zu den heutigen Formen vollzogen hat.
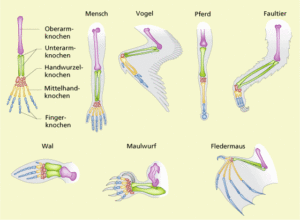
Abb. 3: Die Vordergliedmaßen der Wirbeltiere als Beispiel für das Homologie-Kriterium der Lage. Quelle: https://www.lernhelfer.de/schuelerlexikon/biologie/artikel/homologe-organe
Kriterium der spezifischen Qualität und Struktur
Morphologische (oder andere) Strukturen sind dann zueinander homolog, wenn sie nach dem gleichen Muster gebaut sind, also einen übereinstimmenden bzw. vergleichbaren Feinbau besitzen.
Ein bekanntes Beispiel hierfür ist der Vergleich unserer Zähne mit den Haifisch-Schuppen (sog. Placoidschuppen). Beide haben den selben Feinbau: Die oberste Schicht besteht aus Zahnschmelz und darunter befindet sich das Dentin. Die Gemeinsamkeit des Aufbaus lässt sich damit begründen, dass evolutionsgeschichtlich die urtümlichen kieferlosen Fische über ein Außenskelett verfügten, welches aus Placoidschuppen bestand. Einige dieser Schuppen im Bereich der Mundöffnung hatten sich für die Nahrungsaufnahme spezialisiert und wurden zu Zähnen. Mit der Ausbildung einer knöchernen Wirbelsäule hatte ein stabiles Außenskelett keine Notwendigkeit mehr und wurde reduziert. Übrig geblieben sind lediglich die Zähne und bei den Haien zusätzlich die Placoidschuppen (die eine wichtige Funktion einer effizienten Schwimmbewegung übernehmen).
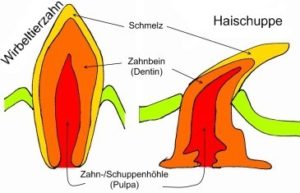
Abb. 4: Zahn und Placoidsschuppe als Beispiel für die Homologie der spezifischen Qualität und Struktur. Quelle: http://www.bio-kompakt.de/images/stories/evolution/zahn.jpg
Kriterium der Kontinuität
Morphologische (oder andere) Strukturen sind dann zueinander homolog, wenn sie durch eine Reihe gleichartiger (homologer) Zwischen- bzw. Übergangsformen miteinander verbunden sind.
Beispiele für das Kriterium der Kontinuität sind z. B. die Kreislaufsysteme der Wirbeltiere oder die Entwicklung der Gehörknöchelchen der Säugetiere, die ursprünglich Teile des Kieferapparates waren.
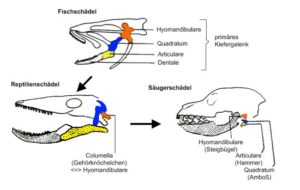
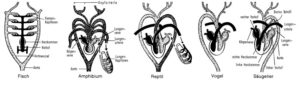
Abb. 5: Kreislaufsystem der Wirbeltiere und die Mittelohrknochen der Säugetiere als Beispiele für das Kriterium der Stetigkeit. Quelle: http://www.bio-kompakt.de/
Von der Homologie klar unterschieden wird die Analogie. Analoge Strukturen sind funktionell gleich, gehen aber nicht auf einen gemeinsamen evolutionären Ursprung zurück. Beispielsweise gehören dazu die Flügel von Insekten und Vögeln oder die Körperformen von Fischen und Delfinen. Sie sind Produkte einer konvergenten Evolution.
Der Vergleich der homologen Organe zeigt schon, dass nicht alle Merkmale für die Bestimmung von evolutionären Verwandtschaftsbeziehungen geeignet sind. Man unterscheidet zwischen Plesiomorphien und Apomorphien. Apomorphien sind abgeleitete Merkmale, also welche die, im Vergleich zum Vorfahren der jeweils betrachteten Stammlinie, neu erworben wurden. Plesiomorphien hingegen sind ursprüngliche Merkmale, die schon die Vorfahren hatten.
Dies kann am Beispiel der Klasse der Säugetiere verständlich gemacht werden (vgl. Abb. 6). Säugetiere müssen, um sie von anderen Gruppen zu unterscheiden, Merkmale aufweisen, die nur ihnen eigen sind, also ihre Apomorphien haben. Hierfür ist beispielsweise die Wirbelsäule ein schlechtes Merkmal, da sie auch bei anderen Wirbeltieren vorkommt. Hierbei handelt es sich also um ein plesiomorphes Merkmal. Hingegen sind Merkmale wie Haare, Schweißdrüsen und die Produktion von Milch typisch für Säugetiere und zwar NUR für Säugetiere. Folgerichtig handelt es sich um apomorphe Merkmale. Die Säugeitere teilt man in zwei Unterklassen ein: die Kloakentiere (Monotremata), wozu u. a. das Schnabeltier gehört und die Zitzentiere (Theria), welche Beuteltiere (Marsupialia) und Plazentatiere (Placentalia) miteinander vereinen. Hier müssen also Merkmale gefunden werden, die nur in einer der beiden Gruppen gefunden werden. Der deutsche Name verrät es schon: Die Zitzentiere (Theria) verfügen über spezialisierte Milchdrüsen und Brustwarzen, während die Kloakentiere nicht nur Eier legen (was erst mal ein plesiomorphes Merkmal ist, da auch Reptilien eierlegend sind), sondern keine Brustwarzen verfügen. Bei ihnen wird die Milch aus der Haut „ausgeschwitzt“ (Milchdrüsen sind nämlich umgewandelte Schweißdrüsen). Will man innerhalb der Theria Plazentatiere und Beuteltiere unterscheiden, so schaut man sich weitere Merkmale an. So verfügen nur die Plazentatiere über eine Plazenta und damit eine „echte“ Schwangerschaft. Die Beuteltiere hingegen verfügen über eine Reduktion der ausgebildeten Milchzähne und das unterscheidet sie von den Plazentatieren. Der namensgebende Beutel hingegen ist keine Apomorphie dieser Gruppe, sondern ist innerhalb der Beuteltiere mehrfach entstanden (Konvergenz), auch besitzen nicht alle Beuteltiere einen Beutel bzw. einige haben ihn reduziert.
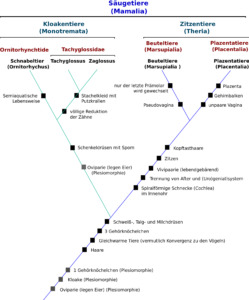
Abb. 6: Typischer Stammbaum der Säugetiere, basierend auf Merkmalsanalysen. Quelle: wikipedia
3. Kladogramme konstruieren
Anhand solcher Merkmalsanalysen, die Phylogenie (Stammesgeschichte) der Taxa widerspiegeln, entstehen entsprechende Diagramme, die man als Kladogramm bezeichnet (in Abb. 1 und 6 für die Primaten bzw. Säugetiere dargestellt).
Da die Merkmale vieler Organismen recht kompliziert sein können und die Listen der verschiedenen Merkmale recht lang sind, wird es ein wenig Kompliziert in diesem Wirrwarr an Merkmalskomplexen einen „richtigen“ Stammbaum zu entwerfen.
Ausgangspunkt einer kladistischen Analyse ist es besonders viele aussagekräftige Merkmale zu finden. Die berücksichtigten Merkmale werden in Form einer Charaktermatrix aufbereitet und dargestellt, in der die Ausprägung des Merkmals durch eine Zahl kodiert wird. Untersucht man die Merkmale einer Gruppe kann man z. B. für das Merkmal „Flügel vorhanden“ mit einer 1 angeben, während „Flügel fehlen“ mit einer 0 angegeben werden.
Idealerweise sollten natürlich schon vorher die Homologien geklärt werden und konvergente Merkmale (Analogie) ausgeschlossen werden. Sind betrachtete Merkmale durch Konvergenzen und zahlreiche unabhängige Entstehungen und Rückbildungen so geprägt, dass sie kaum noch zur Analyse beitragen, spricht man von Homoplasie. Nachdem also die Merkmalsliste erstellt ist, müssen die Apomorphien identifiziert werden. Zur Hilfe bedient man sich sogenannter Außengruppen, um apomorphe und plesiomorphe Merkmale zu unterscheiden. Treten die untersuchten Merkmale auch in diesen Außengruppen auf, so handelt es sich um plesiomorphe, also ursprüngliche Merkmale. Wenn beispielsweise der Stammbaum der Säugetiere untersucht wird, kann man sich zusätzlich die Merkmale einer „Reptilien“-Gruppe als Außengruppe festlegen und wird dabei feststellen, dass eine Wirbelsäule und die Fähigkeit Eier zu legen schon bei dieser Außengruppe vorhanden sind.
Sieht man sich diese Kladogramme an, so wird man sehen, dass es bei jeder Verzweigung immer zwei Äste gibt (dichotome Verzweigung). Jeder dieser Äste weist mindestens eine Autapomorphie auf (also eine Apomorphie, die nur für diesen Ast gültig ist). Die Verzweigungspunkte (Gabelungen) stellen den jüngsten gemeinsamen Vorfahren dar. So gehen Kloakentiere und Theria auf einen gemeinsamen Vorfahren zurück, der sich in diese zwei Linien abspaltete. Da sowohl Kloakentiere als auch Theria auf einen gemeinsamen Vorfahren zurückgehen, also einen Verzweigungspunkt haben, bezeichnet man sie als monophyletisch; sie bilden eine sogenannte Klade, also eine abgeschlossene Abstammungsgemeinschaft. Beide Taxa stellen sogenannte Schwestergruppen dar, die durch gemeinsame Apomorphien (Synapomorphien) gekennzeichnet sind. Und weil Beuteltiere und Plazentatiere ebenfalls einen Verzweigungspunkt haben, sind auch sie monophyletisch und bilden Schwestergruppen. Sie bilden auch sogenannte Kronengruppen, weil sie ganz oben am Baumstamm angelagert sind, an deren Basis der letzte gemeinsame Vorfahr aller rezenten Taxa dieser Klade steht. Die einzelnen Verzweigungspunkte werden jedoch nicht gewichtet, geben also keine Aussage darüber welche Gruppen vorher entstanden sind.
Bei der Aufstellung von Kladogrammen auf Basis solcher monophyletischer Gruppen können, je nach Anzahl der Merkmalen und der untersuchten Taxa, sich mehrere Möglichkeiten ergeben. Dann wählt man diejenige Variante, die mit der geringsten Zahl an Merkmalsänderungen, d. h. Evolutionsschritten auskommt. Man folgt hierbei dem Prinzip der sparsamsten Erklärung, auch Maximum Parsimony genannt. Die Übergänge zwischen einzelnen Merkmalen werden anhand einer Kostenmatrix bewertet und anschließend Bäume mit den minimalsten Kosten für die Übergänge konstruiert. Eine weitere Methodik ist die der größten Wahrscheinlichkeit, auch Maximum Likelihood genannt. Hier wird das Verzweigungsmuster mit der höchsten statistisch errechneten Wahrscheinlichkeit zugrundegelegt. Dazu müssen die Übergangswahrscheinlichkeiten zwischen beobachteten Merkmalen bzw. Sequenzunterschieden ermittelt werden, um den tatsächlichen Umfang evolutiver Schritte einschätzen zu können. Die dritte Methode ist die Neighbor-joining-Methode. Hierbei werden die Abstände evolutionärer Schritte abgeschätzt. Anhand bestimmter Evolutionsmodelle wird die mittlere Anzahl von Veränderungen an einer Position ermittelt, die zwischen einer Sequenz und ihrem Vorläufer oder zwischen 2 Sequenzen aus benachbarten Gruppierungen seit ihrem Divergieren aufgetreten sind. Die Abstände im Sequenzstammbaum werden entsprechend korrigiert. Dies setzt allerdings die Annahme voraus, daß die Zahl an Mutationen in dem vom Sequenzstammbaum erfassten Zeitraum konstant ist. Mit dem sogenannten Bootstrap-Test kann die Verlässlichkeit von Sequenzdaten eingeschätzt werden kann. Dazu wird eine große Zahl von Zufallsdatensätzen generiert, in denen Bereiche des ursprünglich ermittelten Datensatzes wegfallen, andere mehrfach vorliegen, wobei die Länge der Zufallsdatensätze der Länge der ursprünglichen Datensätze entspricht. Dann werden von sämtlichen Zufallsdatensätzen Sequenzstammbäume errechnet und der Anteil festgestellt, mit dem eine bestimmte Gruppierung im Stammbaumdiagramm auftritt. Dieser Bootstrap-Wert steht für die Wahrscheinlichkeit, mit der eine Gruppierung bzw. Verzweigung in der Stammesgeschichte auftritt. Diese Methoden finden vor allem bei Stammbäumen Anwendung, die sich auf molekulare Merkmale stützen (Vergleiche von DNA). Solche molekularen Sequenzanalysen erfordern auch ein sog. Sequenz-alignment. Hierbei handelt es sich um einen Vergleich einzelner Positionen von 2 oder mehr DNA-Sequenzen, mit dem Ziel diese so auszurichten, dass sie in möglichst vielen Positionen identisch oder ähnlich (z.B. Aminosäuren mit ähnlichen Eigenschaften) besetzt sind. Sequenzen mit hoher Ähnlichkeit oder gar Identität stammen mit entsprechend hoher Wahrscheinlichkeit von einer gemeinsamen Vorläufersequenz ab, sind also homolog. All diese Methoden haben ihre Vor- und Nachteile und benötigen teilweise einen großen Rechenaufwand, weswegen auch spezielle Computerprogramme generiert wurden. Entsprechend ist die Fachdisziplin der Bioinformatik, die sich mit der Erfassung, Speicherung, Bearbeitung und Verbreitung biologischer Daten mittels elektronischer Hilfsmittel beschäftigt, von immer größerer Bedeutung.
4. Über „missing links“ und Höherentwicklung
Traditionell wird gerne von einer „Leiter“ der Evolution gesprochen, bei der sich „primitive“ Arten in „höher entwickelte“ verwandeln. Das bekannteste Beispiel stellt die Evolutionsreihe des Menschen vom Australopithecus hin zum Homo sapiens dar. Abb. 7 stellt in lustiger Weise diese Vorstellung dar:

Abb. 7: Vorstellung der Evolution als eine Leiter in Richtung Höherentwicklung.
Anders als ein Stammbaum hat das Kladogramm nur terminale Taxa. Wie an Abbildung 1 oder 6 zu sehen, stehen alle Gruppen am Ende der Verzweigungen. Es lässt damit also nicht die Entwicklung einer rezenten Form aus einer anderen zu. Das heißt: Weder stammt der Mensch vom Schimpansen ab, noch das Plazentatier vom Kloakentier, sondern alle rezenten Arten haben einen gemeinsamen Vorfahren. Anders ausgedrückt: Keine lebende (rezente) Art kann und darf Stammart einer anderen rezenten Art sein. Das hat natürlich Konsequenzen für das Verständnis von Evolution. Denn wenn in einem Kladogramm keine Stammarten gesucht werden ist die Behauptung „Art X“ sei der der Vorfahre von „Art Y“ irreführend. Ein Kladogramm sagt lediglich aus, dass „Art X“ mit „Art Y“ verwandt ist und beide auf einen gemeinsamen Vorfahren zurückgehen, der weder „Art X“ noch „Art Y“ ist. Bei heute lebenden (rezenten) Arten erscheint diese Aussage durchaus plausibel. Dass wir Menschen von den Schimpansen abstammen macht keinen Sinn, da beide Arten heute gemeinsam leben (sofern wir Menschen nicht doch noch auf die bekloppte Idee kommen den Schimpansen auszurotten). Wir beide haben einen gemeinsamen Vorfahren, der weder Schimpanse noch Mensch war, sondern eine eigene Art (der dem Äußeren aber wahrscheinlich dem Schimpansen ähnlicher war als uns). Dies geschah vor etwa 6 – 7 Mio. Jahren und seitdem hatten auch die Schimpansen eine ebenso lange Evolution gehabt wie wir.
Doch was ist mit den ausgestorbenen Arten, die wir als Fossilien kennen? Fossile Arten können in ein Kladogramm integriert werden, sie bilden dann aber ebenfalls terminale Taxa. Das bedeutet, die Zuordnung einer fossilen Art als der tatsächlichen Stammart wird vermieden. Wenn wir also einen Stammbaum des Menschen und der Menschenaffen erstellen, werden wir sehen, dass in unserer „Ahnenreihe“ ein gutes Dutzend verschiedener Hominidenarten entstanden sind. Wir können aber nicht mit Sicherheit sagen, ob Hominiden-Art XY DER VORFAHRE von uns Homo sapiens ist. Warum nicht? Die Erklärung ist ganz einfach: Über 99% aller Lebewesen sind im Verlauf der Erdgeschichte ausgestorben und nur ein winziger Bruchteil davon wird zum Fossil. Und längst nicht jedes Fossil wird auch entdeckt (und es finden sich ja fast permanent neue). Folgerichtig, wenn nicht alle Arten fossilisieren und wir nicht alle Fossilien entdecken, ist es sehr vage zu behaupten Fossil X ist der Vorfahre von rezenter Art Y. Vielleicht kann es der Vorfahre sein, vielleicht aber nur ein entfernter Verwandter. Wir können hierzu auch unseren eigenen Familienstammbaum als Vergleich nehmen. Wir sind das Produkt unserer Eltern. Aber unsere Eltern haben meist Geschwister (unsere Tanten und Onkel), die ebenso Kinder haben, also unsere Cousins und Cousinen. Wiederum kommen noch die Großeltern hinzu, die ebenfalls Geschwister haben. Wenn jemand (z. B. ein Ahnenforscher) nun in meinen Familienstammbaum meinen Cousin, meine Großtante oder meine Urgroßmutter ausfindig machen kann, so ist es offensichtlich, dass sie nicht meine Eltern sein können. Dennoch ist nicht abzustreiten, dass sie mit mir verwandt sind. Genauso verhält es sich mit dem Fossilfund. Wenn wir Fossilien finden und sie verwandtschaftlich in die Nähe rezenter Arten rücken, sagen wir damit nicht, dass wir genau den Vorfahren entdeckt haben, der direkt zur untersuchten rezenten Art führte. Wir sagen lediglich, dass dieses Fossil eine Reihe von Merkmalen mit der rezenten Art hat, die auf eine gemeinsame Abstammung deuten. Es ist, wenn überhaupt, dann ein „Vorfahre“ im weitesten Sinne, sodass wir sagen können wir Menschen stammen von einer Gruppe Primaten ab (weil sich Primaten weit vorher entwickelt hatten, als sich die Wege von Mensch und Schimpanse trennten und der Vorfahre von Mensch und Schimpanse dementsprechend ein Primat war), jedoch nicht von Primatenart XY oder SZ.
Kladogramme zeigen nur die Aufspaltung von Stammarten in Tochterarten (sog. Kladogenese); es werden also Verwandtschaftsbeziehungen aufgestellt. Die Transformation von einem Merkmal zum anderen wird jedoch nicht ermittelt. Dieser Aufgabe widmen sich andere Disziplinen. Die Kladistik widerspricht damit einem „Fortschrittsvorurteil“, das eine Entwicklung „von den Wirbellosen zu den Menschen“ festzustellen meint. Ein grundsätzliches Problem dieser Sichtweise ist, dass dazu der Mensch an die Spitze gestellt werden muss. Tatsächlich steht der „Tendenz“, Wirbel auszubilden, genauso eine „Tendenz“ gegenüber, wirbellos zu bleiben, wie die viel größere Artenvielfalt der Wirbellosen demonstriert. Es gibt also keine höher oder niedriger entwickelten Arten. Da wir alle auf einen gemeinsamen Vorfahren zurückgehen, haben alle heutigen Organismen, von der Bakterie hin zu uns Menschen, die gleiche Evolutionsgeschichte hinter uns. Man hat sich nur in unterschiedliche Richtungen hin entwickelt und spezialisiert. Somit gibt es auch keine „primitiven“ und „höher“ entwickelten Formen, es gibt nur ursprüngliche und abgeleitete Merkmale.
So weisen die Kloakentiere, also eierlegende Säugetiere wie das Schnabeltier, über eine Reihe von ursprünglichen, plesiomorphen Merkmalen auf: es legt Eier, hat eine Kloake, keine Zitzen etc. Gleichzeitig ist es jedoch hoch spezialisiert und weist eine Reihe von Apomorphien auf, die wahrscheinlich frühere, mittlerweile ausgestorbene, Kloakentiere nicht hatten: z. B. das Fehlen von Zähnen, die Giftdrüsen und die spezielle Art der chromosomalen Geschlechtsbestimmung.
Daher können Schnabeltiere – stellvertretend für andere „niedere“ Tierarten – nicht als primitiv angesehen werden. Man spricht eher von basal, weil die erste Abzweigung der Stammlinie der rezenten Säugetiere zu den Kloakentieren führt. Folgerichtig sprechen Kladogramme nicht von einer Hierarchie der Vorfahren, sondern von der Hierarchie der Apomorphien.
Da sich in der Evolution immer neue Arten bilden, neue Spezialisierungen entstehen, von denen keine per se besser ist als die andere, ist das Bild der Evolution einer Stufenleiter von niederen hin zur „Krone der Schöpfung“ falsch. Die Evolution gleicht vielmehr einem Busch, der derselben Wurzeln entspringt, dessen Zweige sich aber in sämtliche Richtungen strecken. Er treibt ständig neue Äste hervor, alte sterben ab. Doch kein Zweig bevorzugt eine bestimmte Richtung.
Folglich ist auch der Mensch nicht das höchste Produkt der Evolution, sondern steht evolutionsbiologisch mit allen anderen Organismen auf dem gleichen Organisationsniveau. Das mag befremdlich wirken, doch wenn wir festhalten, dass einige Bakterien trotz ihrer einfachen Körperbauweise sehr extreme Lebensräume besiedeln, die uns binnen kürzester Zeit umbringen können und sie über Stoffwechselwege verfügen, um Nahrungsquellen zu nutzen, die für uns tödlich sein können, ist die Frage der Höherentwicklung relativ. Auch in ihrer Anzahl sind Bakterien unübertrefflich, da allein ein menschlichen Körper mindestens 100 Billionen Bakterien hat (das ist das 13.000-fache der Weltbevölkerung!). Damit möchte ich die Rolle des Menschen keineswegs herabwürdigen. Auch wir sind eine besondere Spezies, mit besonderen Gehirnen und einem Merkmal, dass bei Tieren allerhöchstens in Ansätzen vorhanden ist: die Gesellschaft. Folgerichtig hat die Biologie keine Monopolstellung auf die Bedeutung des Menschen. Hier haben Gesellschaftswissenschaften, Philosophie und Religion ihr Vetorecht. Doch allein aus der evolutionsbiologischen Perspektive sind wir nicht die Krone des Daseins.
5. Monophylie, Paraphylie, Polyphylie
Wir haben oben festgestellt, dass sich die Kladistik mit monophyletischen Gruppen befasst. Nur diese werden als eine Abstammungsgemeinschaft anerkannt, da sie aus einem gemeinsamen Vorfahren abstammen. Neben monophyletischen Gruppen, gibt es auch paraphyletische und polyphyletische (siehe Abb. 8 – 10). Diese haben folgende Definitionen:
Monophyletisch: Das Taxon hat eine jüngste gemeinsame Stammform (engl. most recent common ancestor, MRCA) und umfasst auch alle Untergruppen, die sich von dieser Stammform herleiten, sowie die Stammform selbst, jedoch keine anderen Gruppen. Das Monophylum begründet sich durch Apomorphien der gemeinsamen Stammform und wird auch als geschlossen bezeichnet.
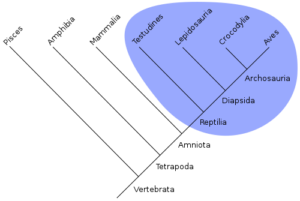
Abb. 8: monophyletische Gruppe der Sauropsida (Erläuterungen siehe Text). Quelle: wikipedia
Paraphyletisch: Das Taxon hat zwar eine jüngste gemeinsame Stammform, enthält aber nicht alle Untergruppen, die auf diese Stammform zurückgehen, wie es beim Monophylum der Fall ist. Ein Paraphylum gründet sich auf die Symplesiomorphien der enthaltenen Taxa und wird auch als offen bezeichnet.
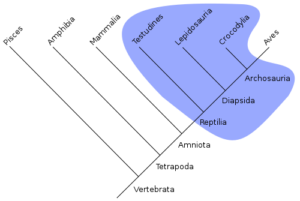
Abb. 9: Paraphyletische Gruppe der Reptilien. Erläuterungen siehe Text. Quelle: wikipedia
Polyphyletisch: Zwei oder mehr Taxa haben gruppendefinierende Ähnlichkeiten ihrer gemeinsamen Merkmale (Morphologie, Funktionen, Struktur von Organen, Verhaltensweisen oder anderer betrachteter Aspekte), die nicht von einem gemeinsamen Vorfahren vererbt wurde, sondern durch konvergente Evolution.

Abb. 10: Polyphyletische Gruppe. Säugetiere und Vögel sind hinsichtlich ihrer Warmblütigkeit Polyphyletisch. Erläuterungen siehe Text. Quelle: wikipedia.
Diese Definitionen haben entscheidende Konsequenzen über unsere Einteilung der Wirbeltiere. In den Abb. 8 – 10 wird jeweils der Stammbaum der Wirbeltiere dargestellt. Wir teilen gerne die Wirbeltiere traditionell in 5 Klassen ein und das schon seit Aristoteles: Fische, Amphibien, Reptilien, Vögel und Säugetiere. Jedoch gibt es mit dieser Einteilung ein entscheidendes Problem: Sie spiegeln keine evolutionsbiologischen Verwandtschaftsverhältnisse wider. Hier sind die Reptilien ein Beispiel für eine paraphyletische Gruppe (Abb. 9). Traditionell zählen wir zu den Reptilien Schlangen, Echsen (im Stammbaum als Lepidosauria dargestellt), Schildkröten (Testudines) und Krokodile (Crocodylia). Bei dieser Einteilung besteht jedoch das Problem, dass z. B. die Krokodile mit den Vögeln viel näher verwandt sind als mit den übrigens Reptiliengruppen. Sie bilden zusammen die monophyletische Gruppe (Klade) der Archosaurier. Hierzu gehören auch die Dinosaurier, die ebenfalls gerne als Reptilien bezeichnet werden. Tatsächlich sind aber einige Dinosauriergruppen mit den Vögeln näher verwandt als mit anderen Dinosauriern. Das heißt übrigens nichts anderes, als dass Vögel Dinosaurier sind! Will man sie nicht als Dinosaurier anerkennen, so müsste das Taxon Dinosaurier in mehrere Gruppen gleichen Ranges aufgeteilt werden. Da jedoch bei den „Reptilien“ die Vögel ausgeschlossen werden, werden nicht alle Nachkommen dieser Stammlinie mit einbezogen. Folgerichtig sind Reptilien in dieser Hinsicht paraphyletisch (siehe Abb. 9). Will man die Reptilien als monophyletische Gruppe darstellen müssen die Vögel mit einbezogen werden. Man bezeichnet diese Gruppe dann aber nicht mehr als Reptilien sondern man spricht von Sauropsida (vgl. Abb. 8). Die Sauropsida bilden zusammen mit den Säugetieren die Gruppe der Amniota. Amniota bezeichnet jene Gruppe der Landwirbeltiere, die hartschalige Eier legen und ihre Fortpflanzung so weitestgehend vom Wasser abgekoppelt haben (und damit haben sie auch kein Larvenstadium). Die Amnioten werden zusammen mit den Amphibien als Tetrapoden (Vierfüßer) bezeichnet. Hierbei handelt es sich um die Landwirbeltiere. Die „Fische“ (Pisces) stellen übrigens ebenso eine problematische Gruppe dar, da einige Fische mit den Landwirbeltieren näher verwandt sind, als mit anderen Fischen. Doch das soll hier nicht vertieft werden.
Die ursprüngliche Gliederung der Wirbeltiere in 5 Klassen wurde eher nach oberflächlichen Ähnlichkeiten zusammengewürfelt (alles, was Schuppen hatte, ist ein Reptil, alles was Kiemen hat und im Wasser lebt, ist ein Fisch) und haben vielleicht ihre didaktische Berechtigung. Sie stellen aber keine realen Verwandtschaftsgruppen dar.
Polyphyletisch sind Gruppen, die keine gemeinsame Stammform haben und keine natürlichen Gruppen darstellen. Ein Beispiel (siehe Abb. 10) ist die Warmblütigkeit. Säugetiere und Vögel sind in der Lage ihre Körpertemperatur unabhängig von der Umgebung konstant zu halten. Dieses Merkmal ist aber das Ergebnis konvergenter Evolution, basiert also nicht auf gemeinsamer Abstammung. Weitere Beispiele für Polyphylie sind solche Bezeichnungen wie „Würmer“, da „Würmer“ phylogenetisch unterschiedliche Stämme darstellen, die nur wenig miteinander gemeinsam haben. Bandwürmer gehören z. B. dem Stamm der Plathelminthes an, der Regenwurm gehört zum Stamm der Anneliden. Damit sind die beiden Gruppen ungefähr so weit voneinander entfernt wie ein Insekt von einer Muschel.
6. Probleme der Kladistik
Die Kladistik oder Phylogenetische Systematik gibt wunderbare Einblicke in die Verwandtschaftsbeziehungen der Lebewesen. Trotz ihrer Vorteile ist sie natürlich nicht frei von Problemen (die sich eventuell mit dem Fortschritten in diesem Bereich lösen lassen). Die Probleme sind in meinem Artikel zur Konstruktionsmorphologie angerissen worden:
- Werden Organismen als Merkmalsträger gewertet, besteht die Gefahr, dass man den Organismus vor lauter Merkmalen nicht mehr erkennt und damit praktisch auflöst. Da der Organismus jedoch in beliebig viele Merkmale aufgelöst werden kann, ist es oft schwer zu ermitteln welches der Merkmale ursprünglich oder abgeleitet ist, bzw. ob das Merkmal auf einen gemeinsamen Vorfahren zurückgeht (also eine Homologie ist) oder nicht. Die Kladistik bringt die festgestellten Merkmale in ein formales Schema. Diese eignen sich zweifelsohne für die Ermittlungen über den Verwandtschaftsgrad, sagen aber noch wenig über Evolutionsprozesse aus. Hierfür muss der Organismus als Gesamtkonstruktion verstanden werden und nicht als bloßer Merkmalsträger.
- Da die Kladistik nur Verwandtschaften zwischen Organismen ermittelt, kann aus ihr nicht gefolgert werden, wie sich einzelne Merkmale im Verlauf der Evolution änderten. Wir haben oben festgestellt, dass die Säugetiere sich in zwei Unterklassen einteilen: die Kloakentiere und die Zitzentiere, letztere wiederrum in Beutel- und Plazentatiere. Doch es wird keine Aussage darüber getroffen, wie sich z. B. der Übergang des Oviparie (legen der Eier) hin zur Entwicklung der Viviparie (lebendgebärend) vollzogen hat. Hier reicht ein reiner Merkmalsvergleich nicht aus, sondern kann bestenfalls Anhaltspunkte liefern. Hier muss die Konstruktion der Organismen verstanden werden, genauso wie die Umweltbedingungen analysiert werden, die diese Veränderungen selektierten. Wenn auch die Kladistik richtigerweise die „Höherentwicklung“ in dem Sinne ablehnt, dass eine basale Art nicht „primitiver“ bzw. „weniger entwickelt“ als eine nicht-basale, spezialisierte Art, so ist es dennoch wichtig zu ermitteln wie sich anatomische (oder verhaltensbiologische und physiologische) Merkmale im Verlauf der Geschichte entwickelten. Im Sinne einer Merkmalsumwandlung, bzw. einer Konstruktionsumwandlung, z. B. die Entstehung der Viviparie der Säugetiere oder die Entwicklung der Flossen und Armen und Beinen), ist eine solche „Höherentwicklung“ berechtigt. Man darf nur nicht den Fehler begehen hierbei rezente Arten (oder auch fossile) als die direkten (primitiven) Vorfahren einer anderen Art anzusehen. Hinzu kommt auch die sogenannten Lesrichtungskritierien. Man muss bei zwei homologen Merkmalen untersuchen, welches denn das ursprünglichere ist.
Man kann das ganze salopp auch so formulieren: durch einen Vaterschaftstest kann man ermitteln, wer der Vater des Kindes ist. Aber der Akt der Zeugung, wie die gesamte Schwangerschaft lässt sich durch einen Vaterschaftstest nicht klären. Genauso muss die Kladistik gesehen werden: wie ein Vaterschaftstest.
- Wenn Organismen nach ihren Merkmalen bewertet werden sollen, besteht die dringende Notwendigkeit die Organismen sehr gut zu kennen. Das Aufstellen von Merkmalen kann nicht oberflächlich analysiert werden, sondern es bedarf einer umfassenden Kenntnis der untersuchten Organismen, da vor allem die zu entdeckenden Apomorphien in vielerlei Hinsicht besonderes Detailwissen erfordern. Das kann bei manchen Gruppen relativ einfach sein, bei anderen wiederrum komplizierter. Gerade bei eher unbekannten oder besonders artenreichen Gruppen können, je nachdem welche und wie viele Merkmale man untersucht und welche Vertreter dieser Gruppen für die Ermittlung der Kladogramme herhalten, verschiedene Kladogramme entstehen, die sich einander widersprechen. Häufig geschieht dies auch, wenn man statt morphologischen molekularbiologische Merkmale nutzt. Gerade bei der Großphylogenie der Metazoa (vielzellige Tiere) entstanden große Diskrepanzen zwischen den morphologischen und molekularbiologischen Stammbäumen.
- Aber gerade die sonst wenig beachtete Konstruktionsmorphologie kann die Probleme, bzw. offenen Fragen, der Kladistik lösen. Das gilt natürlich auch für andere Teilbereiche der Evolutionsbiologie, die sich damit auseinandersetzen, wie bestimmte Strukturen (also Merkmale) entstehen und sich verändern; z. B. die evolutionäre Entwicklungsbiologie oder die Entwicklungsgenetik.