Ein Kerngedanke der Evolution ist, dass alle Arten von Tieren, Pflanzen, Pilzen und Mikroorganismen, die jemals auf der Erde gelebt haben, auf einen gemeinsamen Vorfahren zurückgehen. Das heißt, alle Lebewesen sind miteinander verwandt und lassen sich in einem evolutionären Stammbaum darstellen. Ein solcher Stammbaum stellt die verwandtschaftlichen Beziehungen einer Gruppe von Organismen dar, wie sie nach Darwins Theorie der Abstammung mit Modifikation von gemeinsamen Vorfahren verstanden werden.
Im letzten Beitrag befassten wir uns damit, wie man einen evolutionären Stammbaum richtig lesen kann. In diesem Beitrag wollen wir klären, nach welchen Kriterien diese gemeinsame Abstammung überhaupt geschlossen wird.
Die entscheidende Evidenz für die universelle gemeinsame Abstammung ist die Homologie. Als sich in den frühen 1800er Jahren die vergleichende Anatomie und die vergleichende Embryologie entwickelten, war eines der auffälligsten Ergebnisse, dass den offensichtlichen physischen Unterschieden zwischen den Arten grundlegende Ähnlichkeiten zugrunde liegen. Die frühen Forscher nannten dieses Phänomen Homologie – wörtlich: das Studium der Ähnlichkeit.
Richard Owen, der führende britische Anatom, definierte Homologie als „dasselbe Organ bei verschiedenen Tieren in jeder Form und Funktion“.
Wie kommt es zu diesen Ähnlichkeiten im Aufbau trotz der Unterschiede in Form und Funktion? Darwin argumentierte, dass die Abstammung von einem gemeinsamen Vorfahren die logischste Erklärung ist. Mithilfe von Homologien lassen sich evolutionäre Stammbäume rekonstruieren.
Text als pdf
Die Grundlagen: Taxonomie, Homologe Merkmale
Lebewesen lassen sich auf verschiedene Weisen ordnen. Nehmen wir ein Beispiel aus der Gastronomie, die sog. Meeresfrüchte. Als Meeresfrüchte bezeichnet man in der Regel alle essbaren Meerestiere, die keine Wirbeltiere sind, also Muscheln, Krebse, Tintenfische etc. Hier wird gelinde gesagt alles zusammengefasst was im Wasser lebt, aber kein Fisch ist. Eine solche Einteilung macht für Biologen keinen Sinn, da diese kulinarische Einteilung keine evolutionären Verwandtschaftsverhältnisse widerspiegelt. Auch die Namensgebung ist nicht hilfreich, denn Tintenfische sind eben keine Fische, sondern mit den Muscheln und Schnecken verwandt.
Es braucht also ein Ordnungsprinzip, dass den Evolutionsverlauf und die Verwandtschaftsbeziehungen berücksichtigt. Hierzu habe ich schon eine Reihe von Videos und Texten produziert, die ihr bei Gelegenheit gerne anschauen bzw. lesen könnt. Hier soll aber das wichtigste wiederholt werden:
Die wissenschaftliche Einteilung der Lebewesen wird als Taxonomie bezeichnet. Der früheste Ansatz der Taxonomie stammt vom schwedischen Naturforscher Karl von Linne im 18. Jh., der Lebewesen nach gemeinsamen Merkmalen in ein hierarchisch geschachteltes Ordnungssystem einordnete, bei der jeder untere Rang Teil eines höheren Ranges ist. Linne ordnete die Lebewesen nach anatomischen und physiologischen Merkmalen. So werden ähnliche Arten in Gattungen, ähnliche Gattungen in Familien, ähnliche Familien in Ordnungen, ähnliche Ordnungen in Klassen, ähnliche Klassen in Stämme und ähnliche Stämme in Reiche zusammengefasst (Abb. 1). In der modernen Taxonomie, z. B. bei der Erstellung evolutionärer Stammbäume, haben aber nur noch die Einteilungen „Gattung“ und „Art“ eine Relevanz. Alles darüber nennt man einfach „Taxon“. Das hat z. B. auch damit zu tun, dass zwischen den traditionellen Rängen weitere Ränge möglich sind: So lassen sich Gattungen auch in Untergattungen aufteilen, aber ähnliche Gattungen auch in Gattungsgruppen („Tribus“) zusammenfassen. Dann gäbe es noch Unterfamilien, Überfamilien, Untergattungen usw. usf. Viele dieser Ränge haben noch nicht einmal einen Namen. Das ist die Folge des evolutionären Wandels: Weil alle Lebewesen miteinander verwandt sind, lassen sie sich in detailreiche Gruppen zusammenfassen.
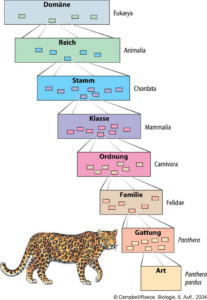
Abb. 1: Taxonomische Ränge
Aber welche Merkmale eignen sich für die Einteilung von Lebewesen? Hier sind homologe Merkmale entscheidend. Gehen Merkmale auf einen gemeinsamen Vorfahren zurück, sind sie zueinander homolog. Das wohl bekannteste Beispiel sind die Vordergliedmaßen bei Wirbeltieren. Obwohl Pferd, Mensch, Vogel, Wal, Maulwurf und Fledermaus ihre Gliedmaßen für völlig unterschiedliche Zwecke verwanden und umgewandelt haben, haben sie dennoch den gleichen Grundaufbau, was auf einen gemeinsamen Vorfahren zurückzuführen ist (Abb. 2).
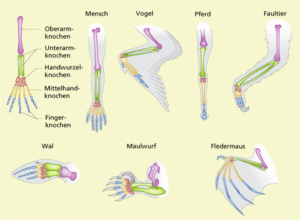
Abb. 2: Homologie der Vordergliedmaßen
Mithilfe von Homologien Stammbäume erstellen
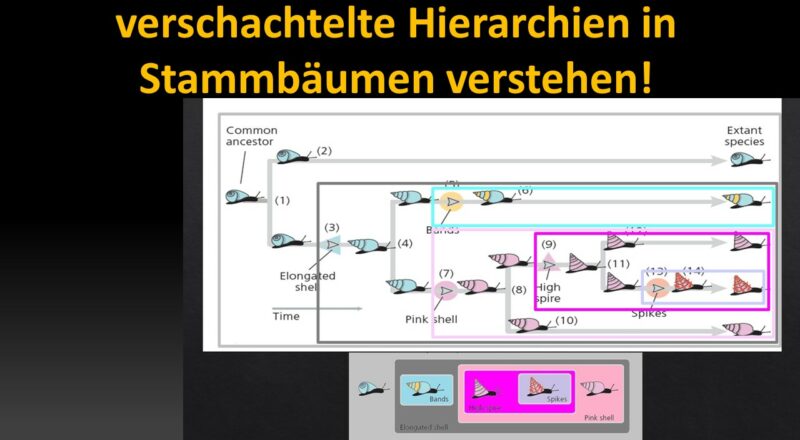
Wir können homologe Merkmale, die die Arten gemeinsam haben, verwenden, um Darwins Hypothese der gemeinsamen Abstammung zu überprüfen. Wir werden die Logik anhand von evolutionären Neuerungen zeigen, die zwischen imaginären Schneckenarten, die sich mit Modifikationen von einer einzigen Abstammungslinie ableiten (Abb. 3).
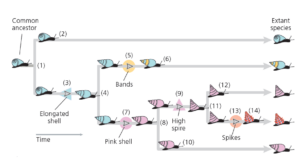
Abb. 3: Stammbaum von fünf imaginären Schneckenarten
Der gemeinsame Vorfahre ist die Linie der blauen Schnecken mit gedrungener Schale ganz links. Es kam dann schließlich zur Artaufspaltung, sodass es zu einer ersten Verzweigung des Stammbaums mit zwei Tochterlinien kommt (1). Eine der Tochterlinien überlebte bis in die Gegenwart, ohne weitere Veränderungen ihrer Schale (2). Die andere Linie entwickelte verlängerte Schalen (3). Die Abstammungslinie mit verlängerten Schalen unterlag einer weiteren Artaufspaltung (4). Eine dieser evolutionären Linie entwickelte Bänder auf ihrer Schale (5), die dann ohne weitere Veränderungen bis in die Gegenwart bestehen blieb (6). Die andere Tochterlinie entwickelte rosa Schalen (7) und spaltete sich dann erneut in zwei Tochterlinien ab (8). Eine Linie entwickelte turmförmige Schalen (9), die andere blieb ohne weitere Veränderungen bestehen (10). Die Linie mit den turmförmigen Schalen spaltete sich ebenfalls in zwei Linien (11). Eine Tochterlinie blieb ohne weitere Veränderungen bestehen (12). Die andere Linie entwickelte Stacheln auf ihrer Schale (13) und blieb dann ohne weitere Veränderungen bestehen (14). Diese Ereignisse führten zu den fünf existierenden Arten ganz rechts.
Jetzt könnte aber folgender Einwurf gemacht werden: Kreationisten werfen gerne vor, dass das Konzept der Homologie eine Tautologie sei: Evolution werde mit Homologie begründet und umgekehrt werde Homologie durch evolutionstheoretische Annahmen bestimmt. Damit entstünde, so die Kritiker, ein Zirkel gegenseitiger Selbstbestätigung, wodurch das evolutionstheoretische Homologieargument an Wert verlöre.
Kreationisten übersehen jedoch, wie perfekt diese Homologien in ein hierarchisch ineinander geschachteltes System gemeinsamer Abstammung passen. Schauen wir uns hierzu nochmal die einzelnen Merkmale der Schnecken an: Man beachte, dass diese Merkmale in einem verschachtelten Muster auftreten (Abb. 4). Die Art mit den Stacheln ist in die Gruppe der Arten mit turmförmigen Schalen eingebettet. Die Gruppe der Arten mit den turmförmigen Schalen ist in der Menge der Arten mit rosa Schalen verschachtelt. Und die Menge der Arten mit rosa Schalen ist in der Menge der Arten mit länglichen Schalen verschachtelt. Aber wir können noch weitergehen. Betrachten wir unser verschachteltes System mit dem vorhin erstellten Stammbaum. Beachtet, dass die am tiefsten verschachtelten Gruppen durch Merkmale wie Stacheln definiert sind, die sich relativ spät entwickelt haben. Wenn wir mit einer dieser Gruppen beginnen und uns über die immer größeren Gruppen, die sie umschließen, vorarbeiten, stoßen wir auf zusätzliche Merkmale, die sich immer früher entwickelt haben. Den Stacheln gingen hohe turmförmige Schalen voraus. Diesen wiederum gingen rosa Schalen voraus. Und den rosafarbenen Schalen gingen die länglichen Schalen voraus.
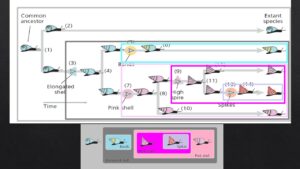
Abb. 4: Verschachtelte Hierarchie der Merkmale der Schneckenarten
Das heißt: selbst wenn man evolutionstheoretische Annahmen nicht beachten würde, würde man auf dasselbe hierarchisch verschachtelte System kommen. Genau das zeigte sich auch bei Linnes System der Taxonomie. Linne war kein Evolutionsbiologe, er glaubte an die Schöpfung und an die Unveränderlichkeit der Arten. Trotzdem ordnete er die Lebewesen in ein geordnetes hierarchisches System. Und es war Linne, der den Menschen in die Familie der Menschenaffen, der Ordnung der Primaten und damit in die Klasse der Säugetiere einordnete.
Selbst wenn wir nur die fünf existierenden Schnecken-Arten hätten und ihre Evolutionsgeschichte nicht kennen würden, könnten wir die Verschachtelung der gemeinsamen Merkmale nutzen, um die Reihenfolge vorhersagen, in der die Merkmale im Fossilbericht erscheinen sollten. Wir könnten dann die Fossilien überprüfen, um zu sehen, ob unsere Vorhersage richtig ist.
Norell und Novacek (1992) haben solche Tests an zwei Dutzend Wirbeltiergruppen durchgeführt. Repräsentative Ergebnisse sind in Abbildung 2.27 dargestellt. In sechs Fällen, z. B. bei Hadrosauriern, gab es keine signifikante Korrelation zwischen der vorhergesagten Reihenfolge, in der die Merkmale auftraten, und der tatsächlichen Reihenfolge (Abbildung 5).
In den anderen 18 Fällen, einschließlich der Reptilien und der Elefanten und ihrer Verwandten, war die Korrelation jedoch signifikant oder stark ausgeprägt.
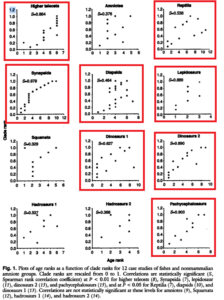
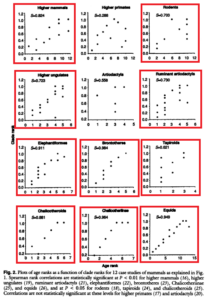
Abb. 5: Ergebnisse der Untersuchungen fossiler Taxa und ihrer Korrelation mit kladistischer Merkmalsverteilung. Die rot umrandeten Graphen zeigen eine Signifikanz beider Parameter.
Bevor jetzt gesagt wird: Moment mal, es gibt also doch Fälle, wo die Evolution nicht stimme. Hier ist Vorsicht geboten: Die biologische Vielfalt ist komplex, der Fossilbericht unvollständig und längst ist noch nicht jede Gruppe ausreichend erforscht. Durch bessere Forschungsmethoden, mehr Datenmaterial usw. können ungelöste, offene Fragen geklärt werden. Das ist ja das Schöne an der Wissenschaft!
Inzwischen wurden ausgefeiltere Methoden zur Bewertung der Übereinstimmung zwischen merkmalsbasierten Rekonstruktionen der Evolutionsgeschichte und der Reihenfolge, in der die Merkmale in den Fossilien auftreten, entwickelt (siehe Wills et al. 2008). Die Übereinstimmung ist im Allgemeinen hoch, zumindest bei gut untersuchten Organismengruppen, die sich gut fossilisieren lassen. Dieses Muster ist mit der Abstammung von gemeinsamen Vorfahren vereinbar.
Homologie wird nicht als Ähnlichkeit aufgrund gemeinsamer Abstammung definiert und dann als Beweis für gemeinsame Abstammung verwendet. Der Beweis für eine gemeinsame Abstammung stammt vielmehr aus den Ähnlichkeitsmustern vieler Merkmale. Diese Ähnlichkeiten zeigen, dass sich Organismen auf natürliche Weise in ein hierarchisch verschachteltes System zusammenschließen. Eine Gruppierung, die von vielen gemeinsamen Merkmalen vorgeschlagen wird, ist ein Beweis für eine gemeinsame Abstammung. Dies gilt unabhängig davon, wie die Merkmale genannt werden.
Es bliebe natürlich immer noch der Einwurf, dass in diesem Ordnungssystem eine göttliche Schöpfung, ein intelligenter Designer etc. stecke.
Aber damit lässt sich das gewünschte Ziel, nämlich die Schöpfungsthese wissenschaftlich zu rechtfertigen, nicht erreichen, weil der Gedankengang methodisch fragwürdig ist. Eine solche Behauptung, dass dahinter ein gemeinsamer Schöpfer steckt, enthält kein wissenschaftliches Argument gegen die Evolution, weil für praktisch alle Fakten auf diese Weise ein Schöpfer postuliert wird. Man kann dann nur noch die Methode der Naturwissenschaft generell infrage stellen und damit den Boden der wissenschaftlichen Argumentation verlassen. Und genau das ist der springende Punkt: Religion oder Glaube ist eben keine Wissenschaft und daher nicht mit wissenschaftlichen Methoden greifbar. Dadurch würde sich jegliche Diskussion um Schöpfung oder Evolution erübrigen, würden Kreationisten nicht versuchen, ihren Schöpfungsmythen ein wissenschaftliches Antlitz zu verleihen.
Hinzu kommt: Wenn es irgendeinen intelligenten Designer gäbe, der alles auf einmal erschaffen hätte, gäbe es keinen Grund, dass er sich an diese Homologiekriterien halten müsste.
So könnte er Fische mit Haaren produzieren, er könnte ein Säugetier erschaffen, welches vier Beine und zwei Paar Flügel hätte und kein Wirbeltier ist oder einen Käfer, der nicht gleichzeitig ein Insekt ist. Man vergleiche z. B. Fische und Delfine miteinander: beide Bautypen haben eine ähnliche Anpassung: einen stromlinienförmigen Körper, um im Wasser zu schwimmen. Aber dennoch ist diese Körperform keine Homologie, sie entstand unabhängig voneinander. Denn wenn wir die Anatomie dieser Tiere genauer untersuchen, merken wir z. B., dass Delfine den Körperbauplan eines Säugetieres haben, ihr Körperbau also homolog zu denen der anderen Säugetiere ist. Delfine haben z. B. Lungen, die dem Bau der Lungen von Säugetieren homolog sind. Warum stattete der intelligente Designer aber Delfine nicht mit einem zusätzlichen Paar Kiemen aus, wie Fische sie haben, wenn er Delfinen schon eine ähnliche Fischform gab? Wenn der Designer doch alle Tiere gleichzeitig erschaffen hat, warum verwendete er das Fischmaterial nicht für die Delfine? Oder warum machte er Delfine nicht gleich zu Fischen?
Natürlich könnte man einwenden, dass ein intelligenter Designer, der dieselben Materialien nutzt, homologe Ähnlichkeiten erschaffen haben könnte. Aber bei der enormen Vielfalt an Lebewesen und der unterschiedlichen Anpassungsstrategien ist es unwahrscheinlich, dass ein intelligenter Designer dahintersteckt. Denn bei jedem erdenklichen Beispiel lässt sich die Homologie, also die gemeinsame evolutionäre Abstammung, beweisen.
Der Mensch ist beispielsweise in die Menschenaffen eingebettet – eine einer Gruppe von Arten, die große Gehirne und keine Schwänze haben. Die Menschenaffen wiederum sind Teil der Primaten, die Greifhände und Füße mit flachen Nägeln statt Krallen haben. Die Primaten sind in die Säugetiere eingebettet, die sich durch Haare definieren und die ihre Jungen mit Milch füttern. Die Säugetiere sind in die Tetrapoden eingebettet, die Tetrapoden innerhalb der Wirbeltiere und so weiter. Das verschachtelte Muster der gemeinsamen Merkmale zwischen den heute lebenden Arten bestätigt somit eine Vorhersage der Darwinschen Theorie.
Nicht zu vergessen: homologe Merkmale sind auch Merkmale, die an die nächste Generation vererbt werden. Durch die Vererbung wissen wir, dass Merkmale einen gemeinsamen Ursprung haben.
Und wir wissen, dass diese vererbten Merkmale variieren. So wie Kinder ihren Eltern ähneln und diese ihren anderen Familienmitgliedern und sich so zu größeren Familienstammbäumen gruppieren ist es genauso mit den Arten. Ähnliche Arten haben ihre Merkmale von anderen ähnlichen Vorläufer-Arten geerbt etc., mit denen sie sich zu größeren Einheiten gruppieren und die verschiedenen systematischen Einheiten bilden.
Dass strukturelle “Information” genetisch an die Nachkommen vererbt wird, ist einleuchtend. Die Vererbung stellt, salopp formuliert, ein “Gedächtnis” dar, “einen Speicher für alle Erfolge, die das Leben jemals errungen hat.”. Wenn also die Merkmale von Generation zu Generation weitervererbt werden, wenn zugleich aber auch ein allmählicher Wandel der Arten stattfindet, dann ergibt sich die logische Folgerung, dass zwischen den Arten eine (abgestufte) Formenähnlichkeit bestehen muss, die sich vom morphologischen bis hinab zum molekularen Bereich erstreckt. So ist für uns die Abstammung offensichtlich, wenn ein Kind seinen Eltern “wie aus dem Gesicht geschnitten” ist.
Entsprechend kann auch an der Verwandtschaft zwischen den verschiedenen Arten kein rational begründeter Zweifel bestehen, wenn man hier auf tiefgreifende Formenähnlichkeiten stößt – vorausgesetzt, man erkennt die Standards wissenschaftlichen Argumentierens an.
Molekulare Homologien
Die strukturellen Übereinstimmungen lassen sich in neuerer Zeit bis in den molekularen Bereich hinein verfolgen, wobei sich zeigt, dass (fast) alle rezenten Lebewesen nicht nur denselben genetischen Code besitzen, sondern auch in weiten Teilen über ein nahezu identisches Repertoire an Genen, Biomolekülen und Stoffwechselprozessen verfügen.
Merkwürdige Ähnlichkeiten, die nichts mit funktionellen Bedürfnissen zu tun haben, treten auch auf molekularer Ebene auf.
Nehmen wir einen genetischen Fehler auf Chromosom 17 beim Menschen. Gemeinsame Fehler sind besonders nützlich bei der Unterscheidung zwischen besonderer Schöpfung und Abstammung von einem gemeinsamen Vorfahren. Der Grund dafür ist jedem Lehrer bekannt, der schon einmal einen Schüler beim Schummeln in einer Prüfung erwischt hat. Wenn A neben B saß und die gleichen richtigen Antworten schrieb, sagt uns das wenig. Wenn aber A neben B saß und identische falsche Antworten schrieb, steigt unser Verdacht. Gleichermaßen lassen gemeinsame Fehler in Organismen auf eine gemeinsame Abstammung schließen.
Der Fehler auf Chromosom 17 befindet sich in der Nähe des Gens für ein Protein namens peripheres Myelinprotein-22 (PMP-22; Abb. 6). Das Gen wird auf beiden Seiten von identischen DNA-Sequenzen flankiert, den so genannten CMT1A-Wiederholungen (Abbildung 2.28a). Diese Situation entstand, als die hintere, distale Wiederholung, die einen Teil des Gens für ein Protein namens COX10 enthält, dupliziert und auf der vorderen, proximalen Seite des PMP-22-Gens eingefügt wurde (Reiter et al. 1997). Diese Wiederholung des vorderen, proximalen CMT1A-Abschnitts war wahrscheinlich Ergebnis eines sog. Crossing-Over während der Meiose. Als Crossing over bezeichnet man einen Austausch von ganzen Chromosomenteilen bei der Meiose. Solche Marker sind gute Indikatoren für eine gemeinsame Abstammung, da es sehr unwahrscienlich ist, dass solche „Fehler“ unabhängig voneinander entstehen (Lopes et al. 1998).
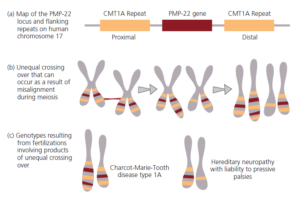
Abb. 6: Ein genetischer Fehler, den der Mensch mit dem Schimpansen teilt (a). Die proximale CMT1A-Wiederholung, in der Nähe des Gens für PMP-22, ist eine Duplikation der distalen Wiederholung auf der anderen Seite des Gens. (b) Die proximale Wiederholung kann sich während der Meiose mit dem distalen Repeat ausrichten, was zu einer ungleichen Überkreuzung führt. (c) Die Genotypen die sich aus der ungleichen Kreuzung resultieren, werden mit neurologischen Störungen verbunden.
Keller et al. (1999) verglichen diese gepaarten CMT1A-Wiederholungen des Menschen mit denen anderer Primaten. Sowohl Schimpansen als auch Bonobos haben mit uns die gepaarten CMT1A-Wiederholungen gemeinsam. Die vordere, proximale Wiederholung fehlt jedoch bei Gorillas, Orang-Utans und allen anderen von den Forschern untersuchten Arten. Dieses Ergebnis ist schwer zu erklären, wenn man davon ausgeht, dass Menschen und Schimpansen getrennt voneinander entstanden sind. Es ergibt jedoch Sinn, wenn man annimmt, dass der Mensch eine Schwesterart der beiden Schimpansen ist. Alle drei Arten haben die proximale Wiederholung von einem gemeinsamen Vorfahren aus jüngerer Zeit geerbt, genau wie drei der Schneckenarten in unserem Beispiel rosa Schalen geerbt haben.
Unser zweites Beispiel für molekulare Homologie betrifft eine andere Art genetischer Eigenart, die als Fehler angesehen werden könnte: prozessierte Pseudogene. Bevor wir erklären, was prozessierte Pseudogene sind, sei darauf hingewiesen, dass die meisten Gene im menschlichen Genom aus kleinen kodierenden Abschnitten, den Exons, bestehen, die durch nicht kodierende Zwischensequenzen, die Introns, getrennt sind. Nachdem ein Gen in mRNA umgeschrieben wurde, müssen die Introns herausgespleißt werden, bevor die Botschaft in Protein übersetzt werden kann.
Beachten wir auch, dass das menschliche Genom mit Retrotransposons übersät ist, retrovirusähnlichen genetischen Elementen, die durch Transkription in RNA, reverse Transkription in DNA und Insertion an einer neuen Stelle im Genom von Ort zu Ort springen (siehe Luning Prak und Kazazian 2000). Einige der Retrotransposons in unserem Genom sind aktiv und kodieren für funktionelle reverse Transkriptase (Abb. 7).
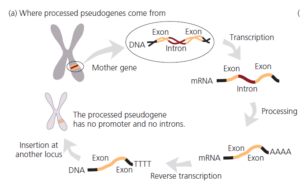
Abb. 7: Prozessierte Pseudogene entstehen, wenn prozessierte Boten-RNAs revers transkribiert und in das Genom eingefügt werden; die Mutationen, die sie angesammelt haben zeigen ihr Alter an.
Nun können wir erklären, dass prozessierte Pseudogene nicht funktionsfähige Kopien normaler Gene sind, die entstehen, wenn prozessierte mRNAs versehentlich von der reversen Transkriptase in DNA umgeschrieben und dann an einer neuen Stelle wieder in das Genom eingefügt werden. Prozessierte Pseudogene sind leicht von ihren Muttergenen zu unterscheiden, da ihnen sowohl Introns als auch Promotoren fehlen.
Eine nützliche Eigenschaft von prozessierten Pseudogenen ist, dass wir ihr Alter schätzen können. Da prozessierte Pseudogene keine Funktion haben, neigen sie dazu, Mutationen zu akkumulieren. Je älter ein prozessiertes Pseudogen ist, desto mehr Mutationen hat es angehäuft. Vergleicht man die Sequenz eines prozessierten Pseudogens mit der seines Muttergens, kann man die Anzahl der Mutationen abschätzen, die das Pseudogen angesammelt hat. Anhand der Mutationen lässt sich das Alter des Pseudogens abschätzen.
Wenn die Arten durch Abstammung von einem gemeinsamen Vorfahren miteinander verwandt sind, dann sollten ältere Pseudogene von einer größeren Vielfalt von Arten geteilt werden. Die Logik hinter dieser Behauptung ist in Abbildung 8 dargestellt. Je früher der Vorfahre, in dem ein prozessiertes Pseudogen auftrat, desto mehr Nachfahren werden es geerbt haben. Einige Nachkommen können das Pseudogen durch Löschung der gesamten Sequenz verloren haben, aber wenn wir genügend Arten untersuchen, sollte das Gesamtmuster klar sein.
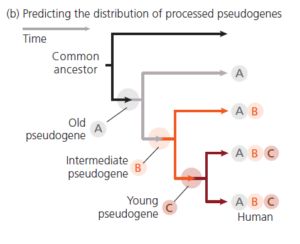
Abb. 8: Wenn Darwins Hypothese der gemeinsamen richtig ist, dann werden ältere prozessierte Pseudogene in einem breiteren Spektrum von Arten vorkommen.
Friedberg und Rhoads (2000) schätzten das Alter von sechs prozessierten Pseudogenen im menschlichen Genom. Das Alter reichte von 11 Millionen Jahren bis 36 Millionen Jahren. Die Forscher suchten dann nach denselben sechs prozessierten Pseudogenen in den Genomen von Schimpanse, Gorilla, Orang-Utan, Rhesusaffe, Kapuzineraffe und Hamster. Die Ergebnisse, die in Abbildung 9 dargestellt sind, stimmen mit unserer Vorhersage überein. Der Mensch hat nur das jüngste der sechs Pseudogene mit den afrikanischen Menschenaffen (Schimpanse und Gorilla) gemeinsam. Die vier Pseudogene mittleren Alters teilen wir mit einer zunehmenden Vielfalt von Primaten (obwohl das 16 Millionen Jahre alte Pseudogen bei Gorillas verloren gegangen zu sein scheint). Schließlich teilen wir das älteste Pseudogen mit den afrikanischen Menschenaffen, den asiatischen Menschenaffen (Orang-Utan), den Altweltaffen (Rhesus) und den Neuweltaffen (Kapuziner). Bei diesen Pseudogenen handelt es sich um molekulare Homologien, deren Verbreitung unter den Primaten ein Beweis für eine gemeinsame Abstammung ist.

Abb. 9: Die Verteilungen der prozessierten Pseudogenen stimmen mit der Vorhersage Darwins überein.
Literatur
Friedberg, F., and A. R. Rhoads. 2000. Calculation and verification of the ages of retroprocessed pseudogenes. Molecular Phylogenetics and Evolution 16: 127–130.
Keller, M. P., B. A. Seifried, and P. F. Chance. 1999. Molecular evolution of the CMT1A-REP region: A human- and chimpanzee-specific repeat. Molecular Biology and Evolution 16: 1019–1026.
Lopes, J., N. Ravisé, et al. 1998. Fine mapping of de novo CMT1A and HNPP rearrangements within CMT1A-REPs evidences two distinct sex-dependent mechanisms and candidate sequences involved in recombination. Human Molecular Genetics 7: 141–148.
Luning Prak, E. T., and H. H. Kazazian, Jr. 2000. Mobile elements and the human genome. Nature Reviews Genetics 1: 135–144.
Norell, M. A., and M. J. Novacek. 1992. The fossil record and evolution: Comparing cladistic and paleontologic evidence for vertebrate history. Science 255: 1690–1693.
Reiter, L. T., T. Murakami, et al. 1997. The human COX10 gene is disrupted during homologous recombination between the 24 kb proximal and distal CMT1A-REPs. Human Molecular Genetics 6: 1595–1603.
Wills, M. A., P. M. Barrett, and J. F. Heathcote. 2008. The modified gap excess ratio (GER*) and the stratigraphic congruence of dinosaur phylogenies. Systematic Biology 57: 891–904.