Proteinbiosynthese
Text als pdf
Die DNA ist das Medium der Zelle, welches die genetische Information für den Aufbau der Proteine speichert. Sie löste damit die RNA ab, die jedoch immer noch eine wichtige Rolle im Zellstoffwechsel spielt. Werden die Informationen aus der DNA in Proteine „übersetzt“, spricht man von der Proteinbiosynthese. Diese Verläuft in zwei Teilprozessen, der Transkription und Translation (Abb. 1). Dabei fließt der Informationsfluss von der DNA zur RNA hin zum Protein. Damit ein Protein-kodierendes Gen exprimiert werden kann, muss es zuerst transkribiert werden. Bei der Transkription wird der Code in der DNA des Gens in einen komplementären Code in einem RNA-Molekül umgewandelt. Diese RNA wird als Messenger-RNA oder kurz mRNA bezeichnet (Munk 2009).
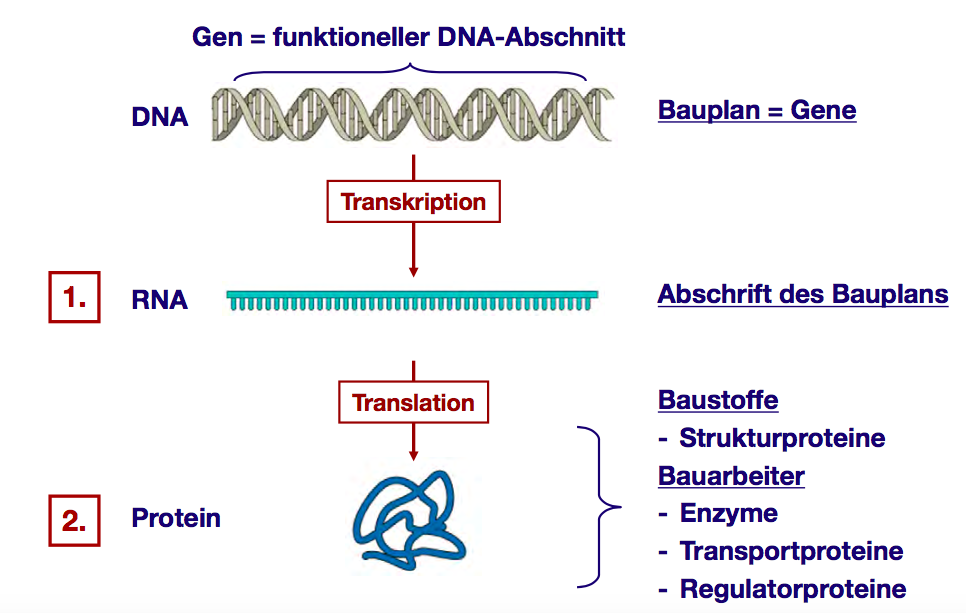
Abb. 1: Vereinfachte Darstellung der Proteinsynthese.
Der genetische Code ist der Schlüssel, mit dessen Hilfe die Nukleotidsequenz einer mRNA, die einem Gen entspricht, in eine Aminosäuresequenz, aus der das von dem Gen codierte Protein besteht, übersetzt (translatiert) wird (Abb. 2). Das heißt, der genetische Code legt fest, welche Aminosäuren nacheinander verwendet werden, um ein bestimmtes Protein zu bilden. Man kann sich den genetischen Code als eine spezielle Folge nicht-überlappender Buchstabenkombinationen der einzelnen Basen der RNA vorstellen. Drei einzelne Nukleotide hintereinander bilden einen Code, quasi die Information für eine Aminosäure. So steht z. B. die Kombination der Buchstaben AAA (3x Adenin) für die Aminosäure Lysin. Der genetische Code ist in den 1960er Jahren entschlüsselt worden. Die damalige Fragestellung war, wie lassen sich mit einem Alphabet, das nur aus vier Buchstaben besteht, mindestens 20 Codewörter schreiben? Oder anders ausgedrückt, wie können die vier RNA-Basen 20 verschiedene Aminosäuren codieren? Da es nur vier Buchstaben gibt, hätte ein Ein-Buchstaben-Code nur vier Aminosäuren codieren können, ein Zwei-Buchstaben-Code nur 4 * 4 = 16. Aber ein Triplettcode mit 4*4*4 = 64 Codons würde mehr als genügen, um 20 Aminosäuren zu codieren. Da es mehr Code-Kombinationen gibt als notwendige Aminosäuren, führt dies dazu, dass manche Aminosäuren mit mehr als einem Code verschlüsselt werden. Der genetische Code ist somit redundant, manche sagen auch „degeneriert“ (Gamow 1954a, b, Crick 1967, Crick et al. 1961, Nirenberg et al. 1965, Judson 1996). Die erwähnte Aminosäure Lysin wird z. B. nicht nur durch die Kombination aus AAA codiert, sondern auch die Kombination AAG. Leucin hat sogar 6 Codes.
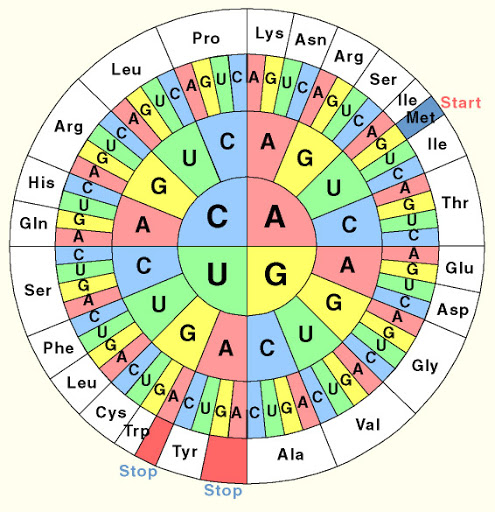
Abb. 2: genetischer Code
Daraus ist übrigens ersichtlich, weshalb eine Rückführung der Aminosäurensequenz zur DNA-Sequenz nicht so einfach ist, da mehrere Aminosäuren für mehrere Basensequenzen stehen können. Von den 64 Codes stehen 61 für Aminosäuren zur Verfügung. Wobei die Aminosäure Methionin durch den Code AUG codiert wird und als Startcodon für die Transkription dient. D. h. jede Proteinsynthese startet mit Methionin als Erkennungssequenz für das Ablesen der Gene. Die drei Codes (UAA, UAG und UGA) sind sog. Stopcodons und beenden die Transkription.
Ist die Transkription abgeschlossen wird in einem zweiten Schritt, der Translation, die mRNA in eine Aminosäure-Sequenz übersetzt. In der Biologie ist dieser Übersetzer eine besondere Art von RNA-Molekül, das man als Transfer-RNA (tRNA) bezeichnet (Abb. 3). Die Translation findet im Cytoplasma an den Ribosomen statt. Für jede der 20 Aminosäuren gibt es mindestens ein spezifisches tRNA-Molekül. Jede tRNA hat ihre Aminosäure im Schlepptau. Sie docken an die komplementäre Stelle der mRNA an, die Aminosäuren werden miteinander verknüpft und fertig ist das Protein.
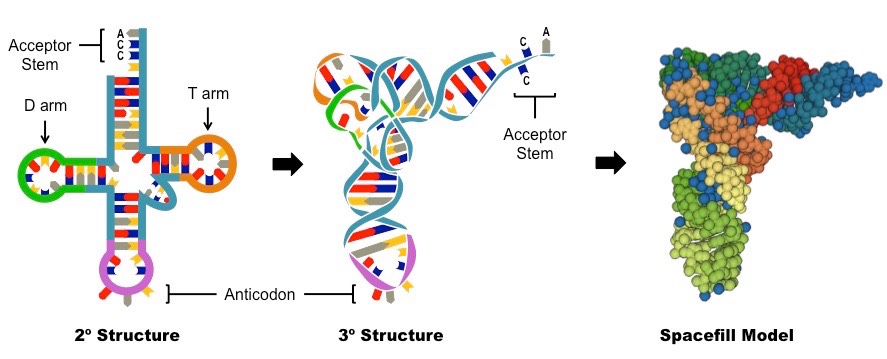
Abb. 3: Struktur der tRNA
Alle Detailschritte der Proteinsynthese, inklusive weiterer Aspekte wie das sog. Splicing, RNA-Editing und die Genregulation wurden von mir schon in anderen Beiträgen vorgestellt[1].
Der Code im Code
Bis auf wenige Ausnahmen verwenden nämlich alle Spezies auf diesem Planeten denselben genetischen Code. Dieser Code wurde in der Evolution des Lebens nahezu durchweg vollständig beibehalten und muss daher immens alt sein. Aber wie ist er überhaupt entstanden (siehe hierzu: Doig 2017, Trifonov 2000, 2004)?
Betrachten wir uns die Verteilung der Aminosäuren in unseren Proteinen, so sind diese mit unterschiedlicher Häufigkeit repräsentiert. Durchschnittlich am häufigsten sind die Aminosäuren Gylcin und Alanin mit jeweils 8-9% vertreten, gefolgt von Leucin (7-8%), Serin (7%), Valin (7%), Glutaminsäure (6-7%) und Asparaginsäure (5-6%). Die Hälfte aller Aminosäuren in unseren Proteinen entfällt also auf diese sieben. Warum ausgerechnet diese? Aus evolutionärer Sicht macht dies Sinn, denn in den Experimenten zur Erzeugung von Aminosäuren unter präbiotischen Bedingungen entstehen diese am häufigsten. Es leuchtet unmittelbar ein, dass diejenigen Biomoleküle, die sich aufgrund physikalisch-chemischer Gesetzmäßigkeiten am häufigsten auf der Erde gebildet haben, auch am ehesten als Bausteine von Lebewesen in Frage kommen. Auch mehr als die Hälfte der Codons des genetischen Codes kodieren für diese sieben Aminosäuren (Neukamm & Kaiser 2014). Damit nicht genug.
Überraschenderweise haben alle Aminosäuren, die aus dem Ausgangsstoff Pyruvat gebildet werden, denselben ersten Buchstaben in ihrem Codon – in diesem Fall ein U. Ist der erste Buchstabe ein C, so leitet sich die verschlüsselte Aminosäure von alpha-Ketoglutarat ab, bei einem A von Oxalacetat. Ist der erste Buchstabe ein G, wird die Aminosäure in einem einzigen entsprechenden Verfahrensschritt aus irgendeiner der anderen einfachen Vorstufen gebildet (Taylor & Coates 1989, Abb. 4). Alle Aminosäurenvorstufen sind Teil des Krebszyklus, welcher sich in den Hydrothermalquellen entwickelt hatte. Der Krebszyklus ist ein zyklischer Stoffwechselprozess, der in den Mitochondrien stattfindet. Er wird gerne als die Drehscheibe des Stoffwechsels bezeichnet, da er eine zentrale Rolle für viele Stoffwechselwege einnimmt. Seine Bedeutung für die Entstehung des Lebens wird in einem gesonderten Beitrag gezielt besprochen.
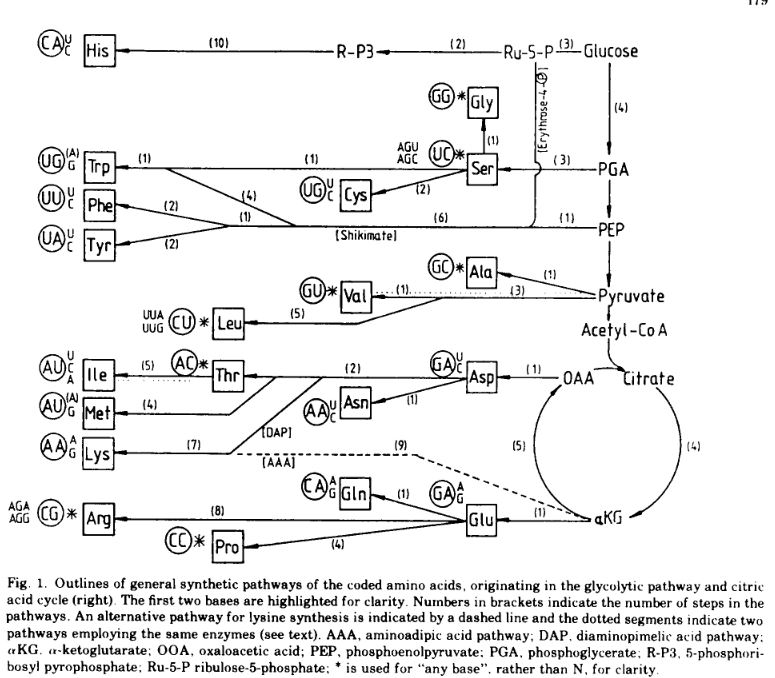
Abb. 4: Der Zusammenhang zwischen dem Krebszyklus und dem genetischen Code. Erläuterungen siehe Text.
Was ist mit dem zweiten Buchstaben? Hier liegt die Information, bis zu welchem Grad eine Aminosäure wasserlöslich ist. Hydrophile Aminosäuren lösen sich im Wasser, hydrophobe in Fetten und Ölen. Fünf der sechs häufigsten hydrophoben Aminosäuren haben ein U als mittlere Base, die häufigsten hydrophilen dagegen ein A, die dazwischenliegenden ein G oder C (Abb. 5). Der letzte Buchstabe ist der Ort, an dem die Degeneration verankert ist – bei acht Aminosäuren eine vierfache Degeneration. Es ist egal welche Base dort sitzt, da alle vier Möglichkeiten dieselbe Aminosäure codieren (Taylor & Coates 1989). Diese Art der Blockzuordnung, sowie die Variabilität der dritten Base legen nahe, dass dem heutigen Triplettcode ein Doppelcode vorausging, der für 16 Aminosäuren codieren konnte (Taylor & Coates 1989).
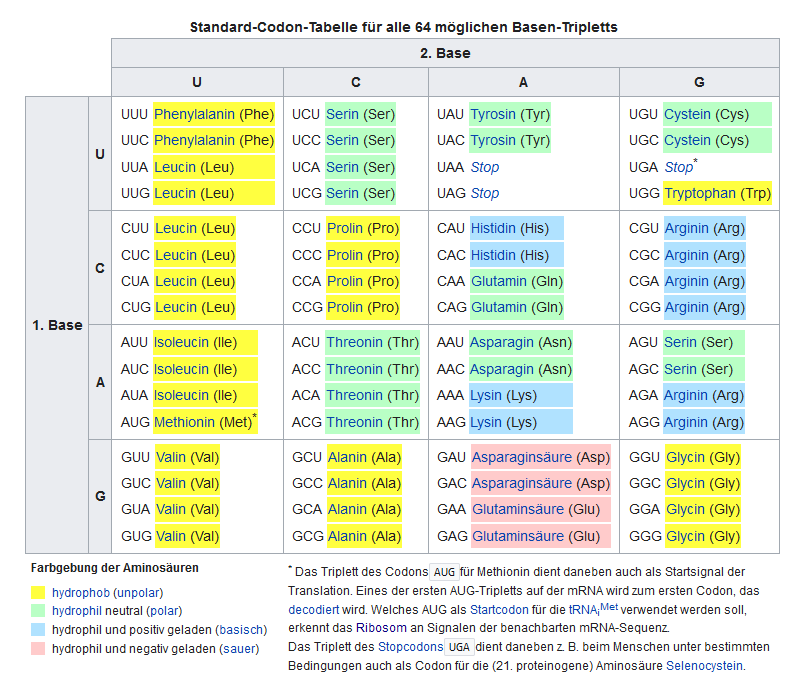
Abb. 5: Eigenschaften den Aminosäuren im Zusammenhang mit dem genetischen Code.
Wenn wir die fünf komplexesten Aminosäuren ausklammern (es bleiben also 15 plus ein Stop-Codon), werden die Muster in den ersten beiden Buchstaben des Codes umso klarer. Es könnte daher sein, dass der ursprüngliche Code ein Doppelcode war und später einfach durch „Codon-Einfang“ zu einem Triplett-Code erweitert wurde (Abb. 6). Die Aminosäuren konkurrieren untereinander um den dritten Platz. Wenn es sich so zugetragen hat, mögen die frühesten Aminosäuren einen unfairen Vorteil was die Übernahme des Triplettcodes angeht gehabt haben. Die 15 Aminosäuren z. B., die höchstwahrscheinlich von dem frühen Doppelcode verschlüsselt wurden, eigneten sich 53 der 64 möglichen Codons an, im Durchschnitt 3,5 Codons pro Aminosäure. Im Gegensatz dazu versammeln die 5 übriggeblieben, späteren Zugänge lediglich 8 Codons unter sich, was einem Durchschnitt von nur 1,6 ergibt. Der frühe Vogel fängt den Wurm (Taylor & Coates 1989).
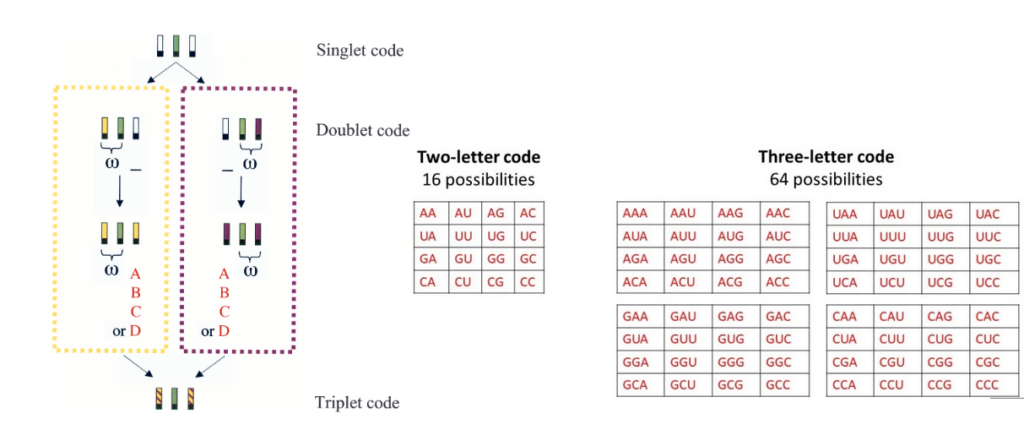
Abb. 6: Doppelcode
Dieser frühe Doppelcode aus 15 Aminosäuren und einem Stoppcodon war offensichtlich durch physikalische und chemische Faktoren vorgeschrieben (Ausgangsstoffe und hydrophile Eigenschaften). Mit dem dritten Buchstaben verhält es sich jedoch anders. Hier war alles so flexibel, dass Spielraum für Zufälle vorhanden war und Selektion dadurch die Möglichkeit hatte, den Code zu optimieren.
Der amerikanische Biochemiker Harold Morowitz und seine Kollegen (Copley et al. 2005) setzten voraus, dass Buchstabenpaare (Dinukleotide) als Katalysatoren fungieren. Sie stellten sich vor, dass sich ein Dinukleotid an die Vorstufe einer Aminosäure, wie beispielsweise Pyruvat, bindet und dessen Umwandlung in eine Aminosäure katalysiert. Welche Aminosäure genau gebildet wird, hängt von den Buchstaben ab, die in einem Dinukleotid gepaart vorliegen. Im Wesentlichen legt der erste Buchstabe den Ausgangsstoff für die Aminosäure fest, der zweite die Art der Umwandlung. Ist das Buchstabenpaar z. B. UU, so formt sich Pyruvat und wird in die ziemlich hydrophobe Aminosäure Leucin umgewandelt (Abb. 7). Um von hier aus zu einem Triplett-Code zu gelangen, sind im Prinzip zwei weitere Schritte nötig, die beide nur eine ganz normale Paarung von zwei Buchstaben voraussetzen. Im ersten Schritt bindet sich ein RNA-Molekül mittels Basenpaarung an das zweibuchstabige Dinukleotid: G an C, A an U etc. Die Aminosäure wird auf die längere RNA übertragen, die stärkere Anziehungskräfte besitzt, je länger sie ist. Das Ergebnis ist eine RNA, die an eine Aminosäure gebunden ist, deren Identität wiederrum von den Buchstaben des Dinukleotids abhängig ist. Im letzten Schritt wird ein Zwei-Buchstaben-Code in einen Drei-Buchstaben-Code überführt, wozu wieder nur eine normale Basenpaarung zwischen RNAs notwendig ist. Wenn solche Wechselwirkungen mit drei Buchstaben besser funktionieren als mit zwei, tritt leichter eine Umschaltung auf einen Triplett-Code ein, in dem die ersten beiden Buchstaben von den Zwängen der Synthese festgelegt werden, während der dritte Buchstabe innerhalb gewisser Grenzen variieren kann, was eine spätere Optimierung des Codes erlauben kann.
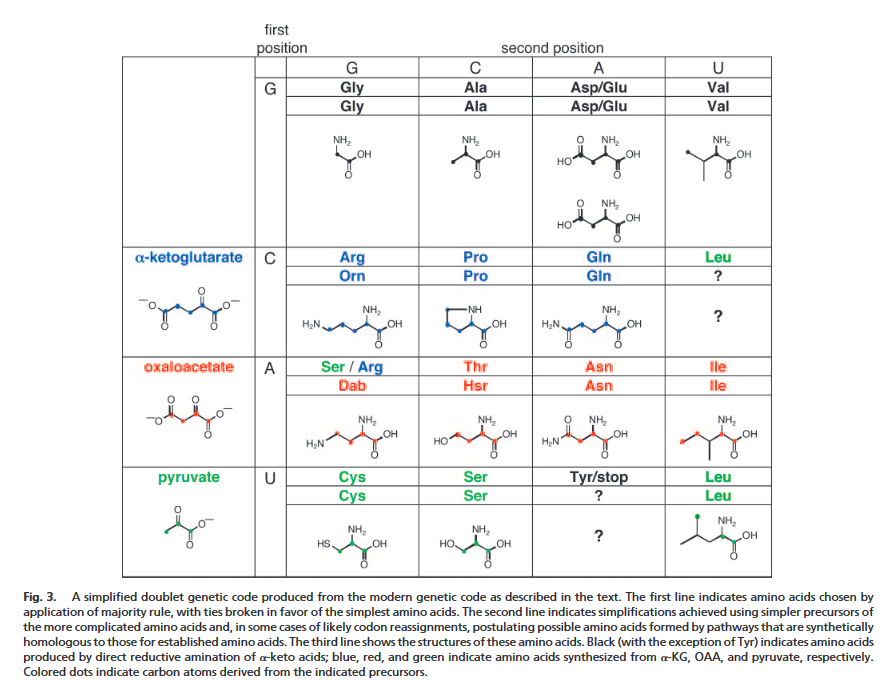
Abb. 7: Ein vereinfachter genetischer Doppelcode, der aus dem modernen genetischen Code, wie im Text beschrieben, erstellt wurde. Die erste Zeile gibt die Aminosäuren an, die durch Anwendung der Mehrheitsregel ausgewählt wurden, wobei die Bindungen zugunsten der einfachsten Aminosäuren aufgebrochen wurden. Die zweite Zeile gibt die Vereinfachungen an, die durch die Verwendung einfacherer Vorläufer der komplizierteren Aminosäuren und in einigen Fällen durch wahrscheinliche Codon-Neuzuweisungen erreicht wurden, wobei mögliche Aminosäuren postuliert wurden, die durch Wege gebildet wurden, die synthetisch homolog zu denen für etablierte Aminosäuren sind. Die dritte Zeile zeigt die Strukturen dieser Aminosäuren. Schwarz (mit Ausnahme von Tyr) zeigt Aminosäuren an, die durch direkte reduktive Aminierung von Alpha-Ketosäuren hergestellt werden; blau, rot und grün zeigen Aminosäuren an, die aus A-KG, OAA und Pyruvat synthetisiert werden. Farbige Punkte zeigen Kohlenstoffatome an, die von den angegebenen Vorläufern stammen.
Einige Wissenschaftler sind sogar der Meinung, dass zum Entstehungszeitpunkt lediglich die mittlere Base eines Codons für die vier häufigsten abiotischen Aminosäuren (Glycin, Alanin, Glutamin- und Asparaginsäure) codierte. Dieser Code hätte sich schrittweise über einen Dublett-Code mit bedeutungsleerer erster oder dritter Codon-Base zum heutigen Triplett-Code expandiert (Wu et al. 2005, Abb. 6 & 8). Die Vorzüge dieses Entstehungsmodells bestehen darin, dass es zwanglos die Nähe von Glutaminsäure und Asparaginsäure sowie von Glutamin und Asparagin und erklären würde, ebenso die Einzel-Codons für Tryptophan und Methionin: Sie zweigten von Isoleucin bzw. von einem Stopp-Codon ab. Dabei werden unvermeidlich ähnliche Codons für chemisch ähnliche Aminosäuren codieren, andernfalls würde die Differenzierung ins Chaos münden. Außerdem würde die mittlere Base als Ursprungsbase die vorhin beschriebene blockweisen Eigenschaften der Aminosäuren in hydrophob, hydrophil und intermediär erklären.
Wu et al. (2005) gehen davon aus, dass ein primordialer Code, in dem lediglich die mittlere Base informationstragend war, zunächst in einen Dublett-Code mit zwei verschiedenen Sorten Codons expandierte: Bei der einen Sorte war die dritte Codon-Base bedeutungsleer (Präfix-Codon), bei der anderen Sorte die erste Codon-Base (Suffix-Codon). Ein solcher Code besäße 2 x 16 = 32 Codons. Als Beleg für ihr Modell führen sie an, dass noch heute Relikte solcher Präfix- und Suffix- Codons zu existieren scheinen, und zwar in Form spezieller Enzyme.
Um die tRNA mit Aminosäuren zu „beladen“, braucht es sogenannte Aminoacyl-tRNA-Synthetasen (kurz: RS, Abb. 9). Eine Besonderheit ist, dass einige RS (ThrRS, ProRS und ValRS) ihre tRNA anhand ihrer ersten beiden Anti-Codon-Basen „erkennen“, andere wiederum anhand der letzten beiden Basen (Abb. 8). Interessanterweise existieren auch RS, die unabhängig vom Anti-Codon ihre tRNA „erkennen“. AlaRS, SerRS und LeuRS nutzen andere Strukturmotive, nämlich die Haarnadelschleife und den „Kleeblattstiel“ der tRNA. Wu et al. (2005) sehen darin einen Beleg dafür, dass der primordiale Code nur für wenige kurzkettige Aminosäuren codierte.
Die Autoren untersuchten nun, was passieren würde, wenn die zwei Sorten Dublett-Codons (Präfix- und Suffix-Codons) zu heutigen Triplett-Codons expandierten: Theoretisch entstünden daraus zwei identische Triplett-Codes. Die praktische Konsequenz wäre, dass die 16 überlappenden Codon-Zuweisungen aus jeder Sorte um Positionen im neuen Triplett-Code konkurrieren würden. Gesetzt den Fall, die Präfix-Codons würden an der dritten Position allen vier möglichen Basen jeweils eine andere Aminosäure (oder Stopp-Signal) zuweisen und selbiges würde spiegelbildlich für die Suffix-Codons gelten, ergäben sich 32 Codierungen (Wu et al. 2005, S. 57.)
Nun hat es einen Selektionsvorteil, wenn überlappende Präfix- und Suffix-Codons für Aminosäuren mit ähnlichen physikalisch-chemischen Eigenschaften codieren. Dies macht die Translation weniger anfällig für Fehler und verhindert, dass die „Zusammenführung“ der überlappenden Dublett-Codons zum heutigen Triplett-Code zu einer „Translations-Krise“ führt. Zudem erweist es sich als vorteilhaft, wenn die Präfix-Codons nach ihrer Expansion an der letzten Basen-Position nicht zwischen allen vier Codon-Basen, sondern nur zwischen Pyrimidin- und Purin-Basen unterscheiden. So codieren zum Beispiel C/U sowie A/G an dritter Basen-Position bei ansonsten gleichen Basen fast immer für dieselbe Aminosäure. Damit ergeben sich statt der 32 nur noch 24 mögliche Codierungen (vgl. Wu et al. 2005, S. 58f.). Somit bietet dieses Entstehungsmodell Raum für etwas weniger als zwei Dutzend Aminosäuren. Das ist genau das, was man beobachtet: Ein Triplett-Code mit 64 Codons und 20 Aminosäuren.
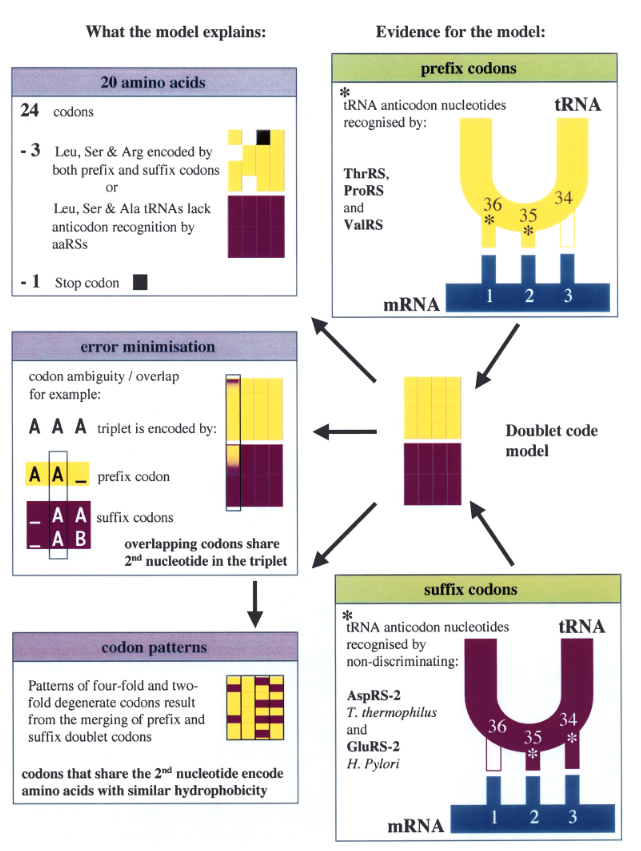
Abb. 8: Die RS für die Aminosäuren Threonin, Prolin und Valin interagieren nur mit den ersten beiden Anti-Codon-Basen (Präfix-Codons). Bei dem gramnegativen Bakterium Thermus thermophilus hingegen existieren RS, die „ihre“ tRNA-Moleküle nur an den letzten beiden Codon-Basen „erkennen“.
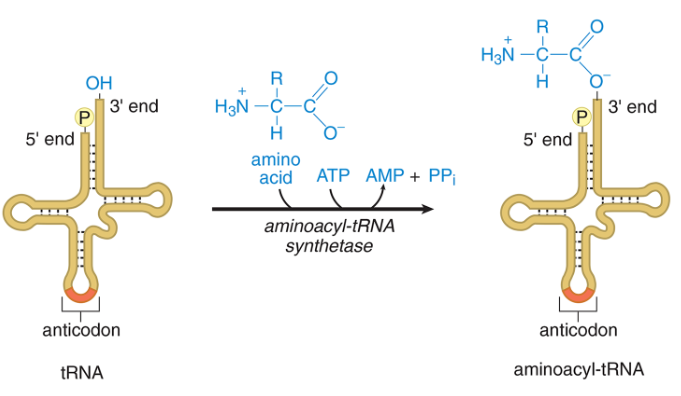
Abb. 9: Aminoacyl-tRNA-Synthetasen
Optimaler Code?
Diese Universalität des genetischen Standardcodes legt nahe, dass er sich schon sehr früh in seiner Geschichte voll ausdifferenzierte und dann „fixiert“ wurde. In den letzten 3,5 Mrd. Jahren scheint er sich nur noch in wenigen Taxa geringfügig verändert zu haben; der genetische Standardcode entspricht also genau jenem Code, den der letzte gemeinsame Vorfahr aller heutigen Lebensformen (LUCA) besaß. Aber wie wahrscheinlich ist es, dass sich genau solch ein Code entwickelt?
In den späten 1990er Jahren verglichen die Molekularbiologen Lawrence Hurst und Stephen Freeland den genetischen Code mit Millionen von zufälligen, am Computer generierten Codes (Freeland & Hurst 1998). Sie berücksichtigten den Schaden, der durch Punktmutationen verursacht werden konnte, indem ein Buchstabe in einem Codon mit indem anderen vertauscht wurde. Sie fanden heraus, dass der echte genetische Code gegenüber genetischen Veränderungen überraschend resistent ist: Punktmutationen behalten oft ihre Aminosäuresequenz bei und im Falle einer Änderung neigen sie dazu, sie durch eine in ihrem Aufbau ähnliche zu ersetzen. Der genetische Code ist demnach besser als eine Mio. alternative, zufällig generierte Codes.
Der genetische Code hat also eine gewisse Optimalität , was jedoch nicht besagt, dass er für diese Zwecke extra „designt“ wäre (wie dies vielleicht Kreationisten behaupten). „Optimalität“ des genetischen Codes bedeutet erstmal Minimierung der Kosten (Neukamm 2021). Das heißt, ein optimaler Code ist maximal „robust“ gegenüber Ablesefehlern und kompensiert die Auswirkungen von Mutationen derart, dass sich die Proteine in ihrer Faltung möglichst wenig ändern. Es gibt hier aber einige Einschränkungen, die zu berücksichtigen sind:
Bislang wurde die Optimalität des Standardcodes üblicherweise hinsichtlich einiger weniger Aminosäure-Eigenschaften untersucht. Der Code soll zum Beispiel robust gegen Änderungen der Polarität der Aminosäuren durch Mutationen sein. Untersuchungen hinsichtlich dieses Aspekts lieferten sicher den überzeugendsten Hinweis auf die Optimalität des Standardcodes.
Die Standard-Methode zur Bestimmung der Optimalität des genetischen Codes hat jedoch einen entscheidenden Haken: Nicht einzelne Aminosäure-Merkmale entscheiden darüber, welche Code-Schlüssel optimal sind. Zahlreiche Eigenschaften beeinflussen das Ergebnis als vernetztes System. Ausschlaggebend sind neben der Polarität der Aminosäuren beispielsweise auch isoelektrischer Punkt, Molekülgewicht, optische Aktivität, Hydrophobizität, Mutabilität, Flexibilität und die Präferenz, bestimmte Sekundärstruktur-Elemente (etwa Beta-Faltblätter) auszubilden (Wnętrzak et al. 2018).
Zweitens beruhen klassische Optimalitäts-Tests auf dem Vergleich des Standardcodes mit zufällig generierten Codes. Der klassische Ansatz, durch zufällig generierte Code-Schlüssel den optimalen Code zu ermitteln, ist nicht erfolgversprechend, da auf diese Weise nur ein verschwindend kleiner Bruchteil aller Aminosäure-Codon-Zuordnungen „erkundet“ werden kann. Neuere Untersuchungen auf der Basis evolutionärer Mehrziel-Optimier-Algorithmen (EMOA) helfen dabei, die beiden oben angesprochenen Probleme zu lösen. 2018 führte eine Arbeitsgruppe (Wnętrzak et al. 2018) unter simultaner Berücksichtigung der acht wesentlichen Aminosäure-Eigenschaften eine solche Studie durch. Im Ergebnis zeigt der Standardcode klare Anzeichen natürlicher Selektion: Er ist zwar deutlich optimiert, jedoch weit davon entfernt, optimal zu sein. Ein beträchtlicher Anteil von Code-Schlüsseln minimiert die negativen Effekte von Aminosäuren-Austauschen hinsichtlich der wesentlichen Aminosäure-Eigenschaften weit deutlicher als der Standardcode (Abb. 10). Mehr noch: Die optimalen Code-Schlüssel unterscheiden sich strukturell erheblich vom Standardcode, was zeigt, dass Optimalität von ganz unterschiedlichen Aminosäure-Code-Zuordnungen aus erreicht werden kann (Koonin & Novozhilov 2009, Koonin 2017).
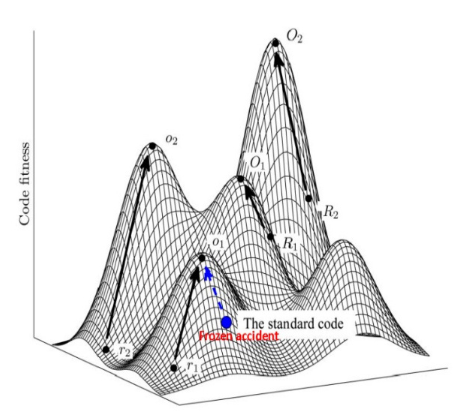
Abb. 10: Die Fitnesslandschaft aller möglichen Code-Schlüssel. Der Standardcode liegt auf einem der „Gipfel“ (o1). Dieses lokale Optimum ist jedoch weit davon entfernt, optimal zu sein.
Eine andere Hypothese besagt, dass die Aminosäuren aufgrund ihrer individuellen chemisch-strukturellen Eigenschaften eine Affinität zu bestimmten Codons aufweisen (Pelc 1965, Pelc & Welton 1966, Dunill 1966, Weber & Lacey 1978, Johnson & Wang 2010, Hou & Schimmel 1988, 1989, Nagamuna et al. 2014). Vertreter dieses sogenannten stereochemischen Modells halten es für plausibel, dass sich der genetische Code zunächst aus einem Ensemble sich selbst vermehrender RNA-Moleküle (sogenannter Ribozyme) organisierte, die direkt bestimmte Aminosäuren binden.
Wie man heute weiß, können bestimmte RNA-Moleküle unmittelbar bestimmte Aminosäuren binden. Diese so genannten Aptamere kodieren also direkt Aminosäuren. So ist es durchaus wahrscheinlich, dass am Ende der RNA-Welt in einem Komplex sich selbst replizierender Ribozyme diverse Moleküle auch einige Aminosäure-Aptamere enthielten. Gesellten sich in der Folge weitere Aptamere hinzu sowie ein Ribozym, welches die Verknüpfung der Aminosäuren zu Oligopeptiden katalysiert, wäre aus einem sich selbst replizierenden Hyperzyklus ein einfacher Translations-Apparat entstanden. Später könnten sich einige dieser Ribozyme auf die Herstellung von Peptiden, andere auf die „Beladung“ von RNA-Molekülen mit Aminosäuren spezialisiert haben, während aus den so beladenen RNA-Molekülen wiederum die verschiedenen Sorten tRNA entstanden sind (Knight & Landweber 2000). Dem Modell zufolge wurden also zu Anfang nicht alle Aminosäuren, sondern zunächst nur einige wenige und dann Schritt für Schritt mehr Aminosäuren von den Organismen genutzt und kodiert (Abb. 11).
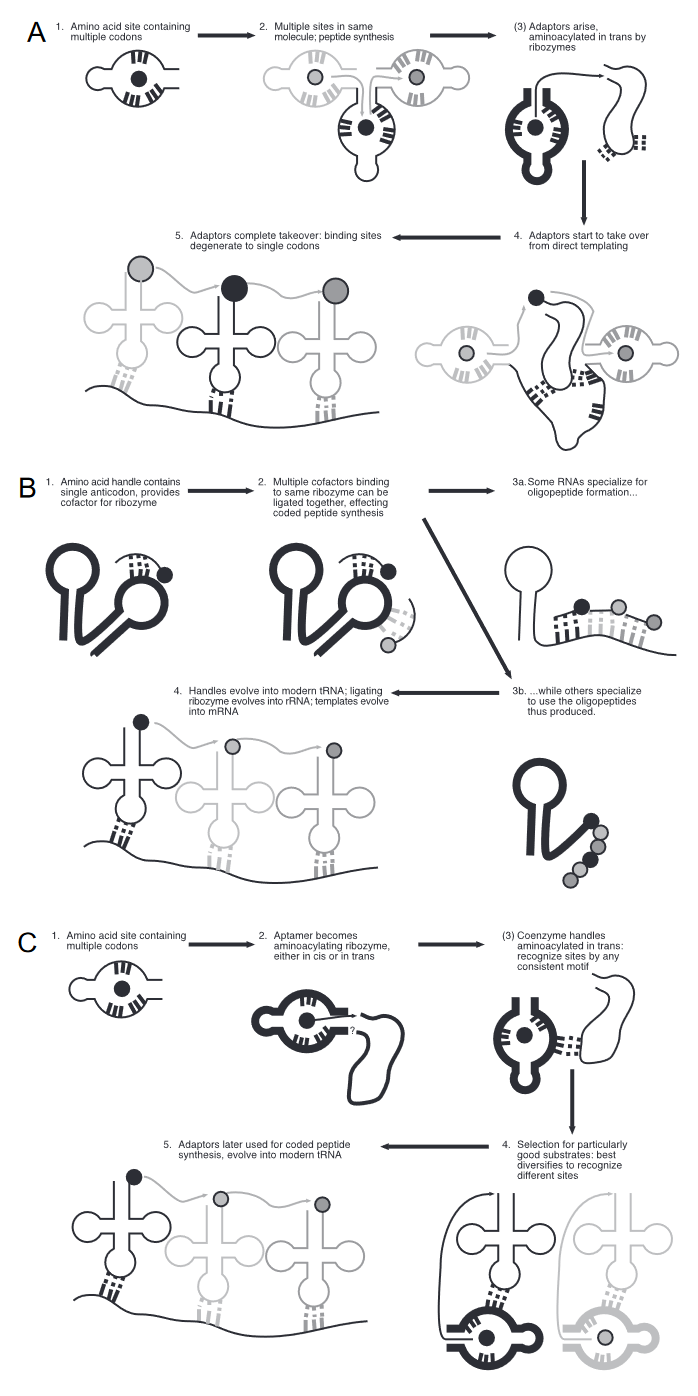
Abb. 11: Vier Modelle zur Entstehung des genetischen Codes: Beim Direct RNA Templating (DRT; Yarus 1998) (A) bilden mehrere Aminosäure-Stellen die ursprünglichen mRNA-Vorlagen; trans-Aminoacylierungs-Ribozyme werden zu moderner rRNA; und Aminosäure-Akzeptoren/Adaptoren werden zu moderner tRNA. Die ursprüngliche Auswahl dient der gerichteten Peptidsynthese. Beim Coding Co-Enzym-Handle (CCH; Szathmáry 1993, 1999) (B) werden Oligonukleotid-Handles, die bestimmten Aminosäuren entsprechen, zur modernen tRNA; einige RNA-Moleküle spezialisieren sich auf die Verwendung dieser Cofaktoren bei der Katalyse und werden zu Ribozymen, die später durch Proteinenzyme ersetzt werden, während andere sich auf die Kodierung spezialisieren und später zu mRNA werden. Die ursprüngliche Selektion dient der nichtkovalenten Bindung von Aminosäuren an Ribozyme. Im Modell von Ellington et al. (2000) entwickeln sich Peptid-ligierende Ribozyme zur Verwendung freier Aminosäuren, die Aptamere entwickeln sich zu modernen tRNA und mRNA. Die ursprüngliche Selektion dient der Bindung positiv geladener Oligopeptide zur Stabilisierung der RNA-Struktur. In der von Knight & Landweber (2000) modifizierten Version von CCH (C) entwickeln sich Aminosäure-Aptamere zu Ribozym-Aminoacyl-tRNA-Synthetasen, die später durch Proteinversionen ersetzt werden; die Aminoacylierungssubstrate entwickeln sich zu modernen tRNA, und mRNA ist eine spätere Erfindung. Die ursprüngliche Selektion ist, wie bei CCH, für die Nutzung von Aminosäuren als Ribozym-Cofaktoren. DRT und modifiziertes CCH stimmen der Beobachtung überein, dass Aptamere Arginin über seine Codons und nicht über seine Anticodons binden.
Computersimulationen (Weberndorfer et al. 2003) konnten solche Modelle plausibel simulieren. Zu Beginn einer Simulation sind lediglich zwei tRNAs definiert, die eine hydrophile und eine hydrophobe Aminosäure tragen. In der simulierten Evolution erweitert sich das Repertoire dann auf bis zu sieben Aminosäuren, für die jeweils eigene tRNAs entstanden sind. Die Triebkraft scheint hier ein besser angepasstes Replikase-Protein zu sein, das bei der Vermehrung der Organismen deren Genom vervielfältigt. Es arbeitet schneller und genauer, wenn es im Verlauf der Evolution aus einer immer größeren Vielfalt von Aminosäuren aufgebaut wird. Am Ende der RNA-Welt mag auf diese Weise der Translations-Apparat und damit der Code entstanden sein: Der Code sollte bereits zu diesem Zeitpunkt fehlertolerant bei Replikation, Transkription und Translation gewesen sein.
Experimentelle Daten, die diese Interpretation stützen, erbrachte eine am Department für Biology und Chemie/Biochemie der University of Colorado ansässige Forschergruppe (Yarus et al. 2009):
Sie rekonstruierten die Struktur RNA-gebundener Aminosäuren und die spezifischen Wechselwirkungen zwischen Aminosäuren und RNA. Die bevorzugt auftretenden Beziehungen wurden dann darauf untersucht, ob die Bindungsstellen Ähnlichkeiten mit dem genetischen Code aufweisen. Es wurde geprüft, ob sie der heutigen Verknüpfung zwischen Aminosäuren und den jeweiligen Anticodons auf der tRNA entsprechen. Dabei ergab sich eine robuste, gegenüber Störfaktoren wenig anfällige Beziehung zwischen den Aminosäuren und den erkennenden Basentripletts in bzw. nahe den analysierten RNA-Bindungsstellen. Die Wahrscheinlichkeit, dass die Basentripletts mit den RNA-Bindungsstellen der Aminosäuren in rein zufälligem Zusammenhang standen, erwies sich als äußerst gering.
Die „Nichtuniversalität“ des genetischen Codes
Dass es eine Evolution des genetischen Codes gab, belegen auch die heute bekannten Abweichungen vom Code. Schon seit 1979 weiß man (Barrell et al. 1979), dass die Abweichungen vom Standardcode vor allem die Mitochondrien der Säugetiere betreffen (Abb. 12). So kodiert in den Mitochondrien das Basentriplett UGA für die Aminosäure Tryptophan, während es im Standardcode die Aufgabe des Stopp-Codons übernimmt. Andererseits sind die Tripletts AGA und AGG in den Mitochondrien Stopp-Signale, während sie im universellen Code für die Aminosäure Arginin stehen. Auch die Ciliaten zeigen Abweichungen vom Standard-Code: UAG, und häufig auch UAA, kodieren für Glutamin; diese Abweichung findet sich auch in einigen Grünalgen.
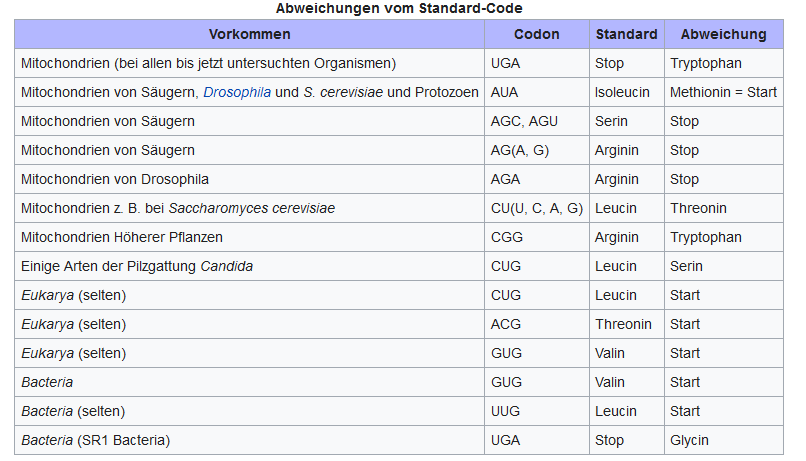
Abb. 12: Abweichungen vom Standardcode
All diese Varianten belegen nicht nur, dass der Code wandelbar ist, sondern geben gleichzeitig Aufschluss darüber, durch welche Mechanismen sich der Code gewandelt haben könnte (Santos et al. 2004). Ausgangspunkt der Abweichungen im Code sind die tRNA-Moleküle (Abb. 13). Eine Mutation in einer tRNA könnte die Beladung der tRNA mit einer bestimmten Aminosäure verändern und damit zu einer (vorübergehend) mehrdeutigen Dekodierung führen. Die mutierte tRNA dekodiert dadurch ein eigentlich „fremdes“ Codon und konkurriert mit der ursprünglichen tRNA. Auf diese Weise könnte auch ein ursprüngliches Stopp-Codon zu einer tRNA beziehungsweise einer zugeordneten Aminosäure kommen.
Man sollte nun meinen, dass solche mehrdeutigen Dekodierungen auf jeden Fall sehr nachteilig für die Organismen sind, weil der Aufbau der Proteine dann nicht mehr zuverlässig geschieht. Tatsächlich aber beobachtet man z. B. in Candida zeylanoides den Fall, dass das normalerweise für Leucin kodierende CUG von einer mutierten Serin-tRNA erkannt wird (Weitze 2006). Diese trägt in rund 95 % der Fälle Serin, ansonsten Leucin. Für den Organismus bringt diese Zweideutigkeit anscheinend keinen wesentlichen Nachteil. Ein Grund dafür ist, dass die Codons von den meisten Lebewesen nicht gleichmäßig genutzt werden. So schwankt die „Nutzungsfrequenz“ der sechs Codons für Leucin zwischen 1 % bis zu annähernd 50 %. Wenn also die tRNA für ein selten genutztes Codon mutiert, kann dies für einzelne Proteine einen Vorteil bringen, während es für die übrigen folgenlos bleibt.
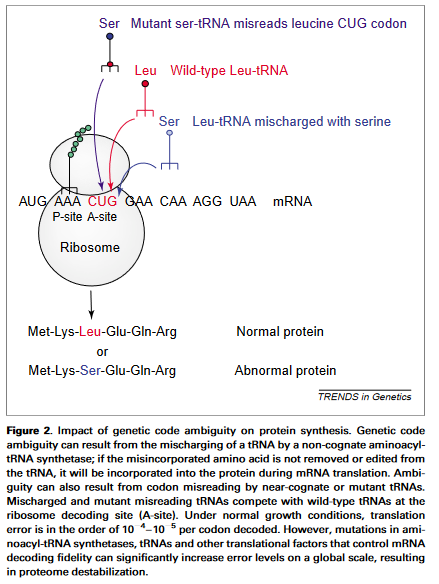
Abb. 13: Auswirkungen der Mehrdeutigkeit des genetischen Codes auf die Proteinsynthese. Die Mehrdeutigkeit des genetischen Codes kann durch die Fehlbeladung einer tRNA durch eine nicht-verwandte Aminoacyl-tRNA-Synthetase entstehen. Wenn die falsch eingebaute Aminosäure nicht aus der tRNA entfernt oder bearbeitet wird, wird sie während der mRNA-Translation in das Protein eingebaut. Mehrdeutigkeit kann auch durch eine Fehlinterpretation von Codons durch nahezu verwandte oder mutierte tRNAs entstehen. Mischarged- und mutierte, falsch lesende tRNAs konkurrieren mit Wildtyp-tRNAs an der Ribosom-Decodierungsstelle (A-Stelle). Unter normalen Wachstumsbedingungen liegt der Translationsfehler in der Größenordnung von 10-4–10-5 pro decodiertem Codon. Mutationen in Aminoacyl-tRNA-Synthetasen, tRNAs und anderen Translationsfaktoren, die die Genauigkeit der mRNA-Decodierung steuern, können jedoch das Fehlerniveau auf globaler Ebene erheblich erhöhen, was zu einer Destabilisierung des Proteoms führt.
Alles in allem scheint der Standardcode das Ergebnis einer partiellen Optimierung eines Zufallscodes zu sein, wonach er sich als robust gegenüber Fehlern bei der Translation erwies. Der Grund dafür, dass der Code nicht vollends optimiert wurde, scheint ein relativ einfacher zu sein: Der wachsende Konflikt zwischen dem vorteilhaften Effekt dieser zunehmenden Robustheit und dem schädlichen Effekt der reihenweisen Umprogrammierung der Codons, der mit der wachsenden Komplexität des evolvierenden Systems immer schwerwiegender wurde (Novozhilov et al. 2007). Die Evolution des Codes kann damit als eine Kombination von Anpassung und „frozen accident“ angesehen werden (Crick 1967, Sella & Ardell 2006). Andere Autoren möchten den „frozen accident“ allerdings lieber als „aufgetaut“ ansehen (Söll & RajBhandary 2006).
Evolution der RNA-Polymerase
Bei der Proteinbiosynthese spielen Enzyme wie die RNA-Polymerase eine wichtige Rolle. RNA-Polymerasen wirken bei der Synthese einer Ribonukleinsäure (RNA) aus ihren Grundbausteinen (Ribonukleotide) mit (Abb. 14). Je nachdem ob die Polymerasen eine RNA oder DNA als Matrize nehmen, spricht man von RNA bzw. DNA-abhängigen RNA-Polymerasen (Munk 2009).
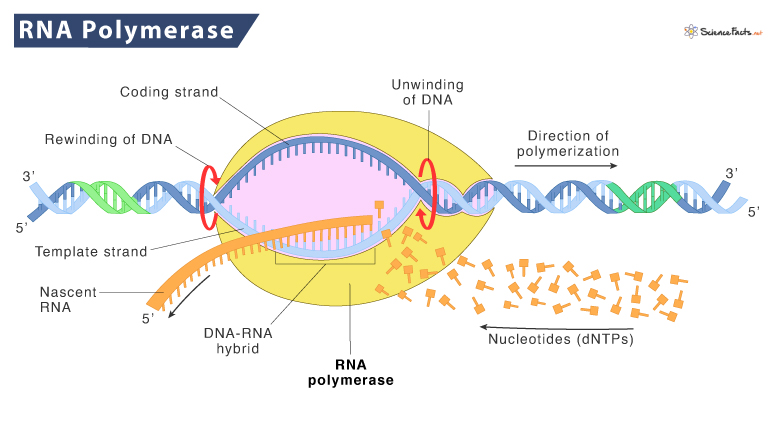
Abb. 14: allgemeine Funktion der RNA-Polymerase
RNA-abhängige RNA-Polymerasen, auch als RNA-Replikase bezeichnet, kommen hauptsächlich in Viren vor. In eukaryotischen Zellen (von Tieren, Pflanzen und Pilzen) dienen RNA-Replikasen dagegen im Zuge der RNA-Interferenz dem Aufbau doppelsträngiger RNA. Die RNA-Interferenz ist ein natürlicher Mechanismus, welcher der zielgerichteten Abschaltung von Genen dient. Bei Bakterien gibt es eine DNA-abhängige RNA-Polymerase, die an der Transkription der Gene und Synthese der mRNA beteiligt ist. Eine weitere ist bei der Replikation beteiligt, nämlich jene, die die RNA-Primer, die zum Start der Replikation genutzt werden, synthetisiert. Bei Eukaryoten gibt es mehrere DNA-abhängige RNA-Polymerasen, die alle miteinander und zu jener der Bakterien homolog sind. RNA-Polymerase I synthetisiert eine rRNA, RNA-Polymerase II die mRNA und RNA-Polymerase III die tRNA. RNA-Polymerasen IV und V sind an der Synthese der siRNA beteiligt (Abb. 15).

Abb. 15: eukaryotische RNA-Polymerasen
Die evolutionär ältesten RNA-Polymerasen sind die RNA-abhängigen RNA-Polymerasen und ein Relikt der RNA-Welt (de Farias et al. 2017). Der Ursprung RNA-abhängigen RNA-Polymerasen kann auf tRNA-Vorfahren zurückverfolgt werden, die bei der Entstehung des primitiven Übersetzungssystems, bei dem die RNA das Informationsmolekül war, als erste Gene fungierten. Der älteste Teil des Enzyms ist die Cofaktor-Bindungsstelle, die in der Lage war, einfache Moleküle wie Magnesium, Kalzium und Ribonukleotide zu binden.
Die Evolution der RNA-abhängigen RNA-Polymerasen ist untrennbar mit der Evolution von RNA-Viren verbunden, vor allem weil dieses Enzym für die Replikation der Genome von RNA-Viren verantwortlich ist. Dies legt auch den Schluss nahe, dass RNA-Viren evolutionsbiologisch sehr alt sind (Černý et al., 2014; Jácome et al., 2015).
Die evolutionäre Position von RNA-Viren im Stammbaum des Lebens ist ein kontroverses Thema in der modernen Biologie (Jácome et al., 2015; Nasir & Caetano-Anollés, 2015). Ihre Häufigkeit als Parasiten eukaryontischer Organismen deutet auf einen jüngeren Ursprung hin, aber Arbeiten, die die Evolution der Protein-Superfamilie analysiert haben, legen einen sehr alten Ursprung nahe, da sie Zeitgenossen des letzten universellen gemeinsamen Vorfahren sind (Nasir & Caetano-Anollés, 2015). Die Unverzichtbarkeit der RNA-abhängigen RNA-Polymerase in RNA-Viren sowie in der RNA-Welt wirft Fragen und Szenarien zu einem jüngeren Ursprung dieser Viren auf. In einer RNA-Welt ist das Auftreten einer RNA-abhängigen RNA-Polymerase ein wesentlicher Schritt für die Entstehung eines ursprünglichen Genoms auf RNA-Basis.
Basierend auf den vorliegenden Ergebnissen kann ein hypothetisches Szenario vorgeschlagen werden, bei dem ein Ribozym mit Polymeraseaktivität seine Aktivität durch die Bindung eines einfachen Cofaktors wie Magnesium (Shechner et al., 2009; Horning & Joyce, 2017) erhöht haben könnte und so die Funktionen der Replikation einiger auf RNA-Molekülen gespeicherter Informationen ausübt (Kim & Higgs, 2016; Tagami et al., 2017). Mit der Entstehung der ursprünglichen Transkription wurden die ersten Proteine durch Übersetzung der Verbindung von proto-tRNAs gebildet, und die RNa-abhängige RNA-Polymerase war eines der ersten Enzyme, die gebildet wurden (Farias et al., 2016). Begleitet vom Prozess der Kompartimentierung konnte die Kontrolle der gelösten Stoffe in der internen Umgebung und die Polymerisationsfunktion durch Proteine effizienter erfolgen als bei Ribozymen. Ursprünglich bestand das Enzym nur aus der katalytischen Schleife mit der Fähigkeit zur Bindung an den Cofaktor und einfache Moleküle wie Ribonukleotide. Durch Duplikationen und Diversifikationen der ursprünglichen katalytischen Domäne konnten die anderen Teile des Proteins hervorgehen. Mit der Etablierung der ersten Domäne konnten Varianten erzeugt werden und Mutationen auftreten, so dass es möglich war, neue Proteine zu erzeugen, wobei die katalytische Stelle erhalten blieb (Zong et al., 2009). Mit der Entstehung einer Variante mit den Eigenschaften einer reversen Transkriptase und später einer DNA-Polymerase erfolgte ein grundlegender Schritt zur Entstehung der ersten Genome auf DNA-Basis, der die Brücke von einer RNA/Protein-Welt zu einer DNA/RNA/Protein-Welt schlug (Gilbert, 1986; Müller, 2006). Als die DNA-Moleküle auftauchten, traten durch Variation andere Klassen von Polymerasen auf. So haben wir in der letzten Episode kennengelernt, dass die DNS-Polymerase D, die in einigen Archaeen vorkommt, selbst aus einer RNA-Polymerase hervorgegangen ist.
Eukaryotische DNA-abhängige RNA-Polymerasen (Pol I-III) haben einen konservierten Kern aus 12 Untereinheiten, der eng mit archaeischen RNA-Polymerasen verwandt ist (Kwapisz et al. 2008, Abb. 16). Früher wurde angenommen, dass Rpb8, eine Untereinheit der RNA-Polymerasen I, II und III, auf Eukaryoten beschränkt ist. Diese Untereinheit hat jedoch hohe Sequenzähnlichkeiten mit einer Untereinheit der RNA-Polymerasen der Gruppe der Crenarchaea, einem Protein namens G, wodurch ein letztes fehlendes Bindeglied zwischen der Kernstruktur archaeischer und eukaryotischer RNA-Polymerasen identifiziert wird.
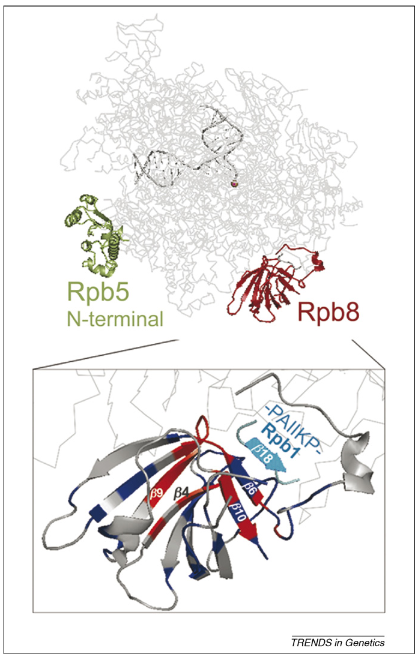
Abb. 16: Sequenz von Rpb8/G und seiner Rpb1-b18-Verankerungsstelle in Rpb1. (a) Rpb8/G-Ausrichtungen, dargestellt für die Crenarchaea. Invariante oder hochkonservierte Positionen sind rot dargestellt. (b) Lokale Ausrichtungen von Rpb1 (Pol II), Rpa190 (Pol I), Rpc160 (Pol III) im Vergleich zu ausgewählten archaischen und DNA-Virus-Sequenzen. Der Rpb1-b18-Konsensus und die entsprechenden Artennamen sind rot dargestellt.
Zwei Szenarien sind hier denkbar: Erstens, alle eukaryotischen und archaeischen RNA-Polymerasen haben einen gemeinsamen Ursprung mit der Rpb8/G-Untereinheit und diese ist bei den anderen Kladen der Archaeen verlorenen gegangen. Zweitens wäre es aber auch denkbar, dass das G-Protein von den Crenarchaea erworben wurde, nachdem sie sich von den anderen Archaeen abgespalten haben. Dies bedeutet, dass das eukaryotische Kerngenom von Crenarchaea-ähnlichen Vorfahren abstammt, die die G-Untereinheit in ihrer RNA-Polymerase enthielten (Abb. 17).
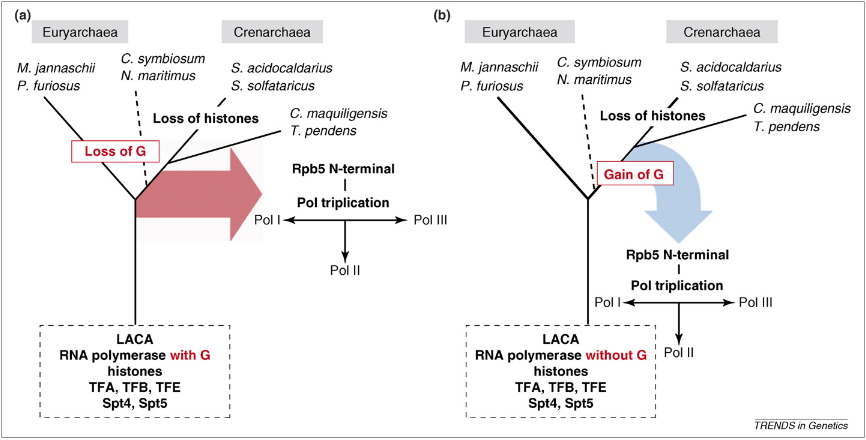
Abb. 17: Zwei Szenarien für die Entwicklung von DNA-abhängigen RNA-Polymerasen bei Archaeen und Eukaryoten. Eine RNA-Polymerase der Vorfahren mit Homologie zur gesamten Untereinheiten-Kernstruktur, die Pol I, II und III gemeinsam ist, einschließlich der G-Untereinheit, war wahrscheinlich im letzten gemeinsamen Vorfahren der Archaeen (LACA) vorhanden. Dieses Enzym könnte auf DNA-Templates eingewirkt haben, die Archaeen-Histone enthielten, und mit drei Initiationsfaktoren (TFA, TFB und TFE in der Archaeen-Nomenklatur) und mit mutmaßlichen Elongationsfaktoren, die Spt4 und Spt5 ähneln, interagiert haben. Histone gingen bei den meisten Crenarchaeen-Phyla verloren, blieben aber bei Arten erhalten, die mit der heutigen C. maquiligensis/T. pendens-Gruppe verwandt sind. Die frühe Evolution eukaryotischer RNA-Polymerasen beinhaltete die Übernahme der N-terminalen Domäne von Rpb5 und eine Verdreifachung, die zu Pol I, II und III führte. Alternativ könnten diese drei Enzyme auch verschiedene archaische Vorfahren haben. Eine gestrichelte Linie kennzeichnet die kaum erforschte Phylogenie der C. symbiosum/N. maritimus-Gruppe. (a) Hypothetisches Szenario, das auf einer frühen Entstehung (hier durch einen roten Pfeil symbolisiert) des eukaryotischen Kerns aus LACA-ähnlichen Vorfahren der Archaeen basiert. Die G-Untereinheit von LACA ging bei Euryarchaea und in der Gruppe C. symbiosum/N. maritimus verloren. Breite rote und blaue Pfeile symbolisieren den noch wenig erforschten Prozess, vermutlich symbiotischer Natur, der letztlich den ursprünglichen eukaryotischen Kern hervorbrachte, zumindest teilweise ausgehend von DNA archaischen Ursprungs. (b) Hypothetisches Szenario, das auf einer späten Entstehung (hier durch einen blauen Pfeil symbolisiert) des eukaryotischen Kerns aus mit Histonen ausgestatteten crenarchaischen Vorfahren basiert. G wäre im LACA-Enzym nicht vorhanden gewesen und wäre speziell in Crenarchaea erworben worden.
Evolution der tRNA
Wenn die Evolution der RNA-abhängigen RNA-Polymerasen auf tRNA-Vorfahren zurückverfolgt werden kann, lohnt sich ein Blick auf die Ursprünge der tRNA. Nachdem die mRNA synthetisiert wurde, lagert sich diese an die Ribosomen und wird ihrerseits von den tRNAs abgelesen. Jede tRNA hat eine spezifische Aminosäure im Schlepptau und die tRNA verknüpft sich an die komplementären Basen der mRNA. Die tRNA wurde 1958 von Mahlon Bush Hoagland und seiner Arbeitsgruppe nachgewiesen (Hoagland et al. 1958). Eine typische tRNA besteht aus 76 Nukleotiden, es können aber auch mehr sein. Die klassische Sekundärstruktur, die gewöhnlich als „Kleeblattstruktur“ bezeichnet wird, besteht aus einem CCA-Ende an Position 74-76, einem Akzeptorstamm an Position 1-7 und 67-73, einer Anticodon-Schleife an Position 30-46, einer T-Schleife an Position 52-68, einer D-Schleife an Position 8-24 und einer variablen Schleife (Abb. 3 und 18).
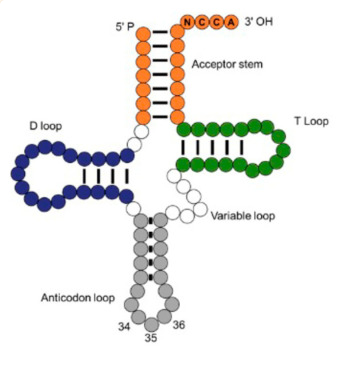
Abb. 18: tRNA
Obwohl die meisten tRNAs die klassische Struktur aufweisen, gibt es hier einige Abweichungen, z. B. in Mitochondrien (Ohtsuki et al. 2002, Masta & Boore 2008, Ohtsuki & Watabane 2007). Im gefalteten Zustand nimmt die tRNA eine nahezu senkrechte Winkelgeometrie an und Magnesium-Ionen spielen eine Rolle bei der Stabilisierung der tRNA-Geometrie (Jovine et al. 2000).
Wie ist die Struktur der tRNA entstanden? Zur Entstehung der Struktur der tRNA gibt es mehrere Hypothesen. Im Wesentlichen gehen sie davon aus, dass verschiedene Sekundärstrukturen, z. B. sog. Haarnadel-Strukturen, sich bildeten und untereinander oder mit Aminosäuren interagieren bzw. sich selbst als Vorlage für die Replikation weiterer RNA-Stränge nutzen, die sich miteinander verbanden (Woese 1969, Hopfield 1978, Eigen & Winkler-Oswatitsch 1981, Bloch et al. 1985, Möller & Janssen 1992, Di Giulio 1992, 2004, 2020, Widman et al. 2005, Tanaka & Kikuchi 2001, Nagaswamy & Fox 2003, Maizels & Weiner 1994, 1999, Harvey & McCammon 1981, Demongeot & Moreira 2007, Demongeot & Seligmann 2019, Sun & Caetano-Anollés 2008, Rodin et al. 2011, Petrov et al. 2015, Burton 2020, Pak et al. 2017, Kühnlein et al. 2021, Ludwig 2021, Wu et al. 2022; Zusammenfassung bei Guo & Su 2022, Abb. 19 & 20). Die obere Hälfte der tRNA (bestehend aus der T-Schleife und dem Akzeptorstamm mit 5′-terminaler Phosphatgruppe und 3′-terminaler CCA-Gruppe) und die untere Hälfte (bestehend aus der D-Schleife und dem Anticodon) sind sowohl in der Struktur als auch in der Funktion unabhängige Einheiten. Die obere Hälfte könnte sich zuerst entwickelt haben und die 3′-terminale genomische Markierung enthalten, die ursprünglich tRNA-ähnliche Moleküle für die Replikation in der frühen RNA-Welt markiert haben könnte. Die untere Hälfte könnte sich später als Erweiterung entwickelt haben, z. B. als die Proteinsynthese in der RNA-Welt begann und sie in eine Ribonukleoprotein-Welt (RNP-Welt) verwandelte. Dieses vorgeschlagene Szenario wird als Genomic-Tag-Hypothese bezeichnet. Tatsächlich haben tRNA und tRNA-ähnliche Aggregate auch heute noch einen wichtigen katalytischen Einfluss (d. h. als Ribozyme) auf die Replikation. Diese Rollen können als molekulare (oder chemische) Fossilien der RNA-Welt betrachtet werden (Maizels & Weiner 1999). Im März 2021 berichteten Forscher über Hinweise, die nahelegen, dass eine frühe Form der Transfer-RNA ein Replikations-Ribozym-Molekül in der sehr frühen Entwicklung des Lebens gewesen sein könnte (Kühnlein et al. 2021, Ludwig 2021).
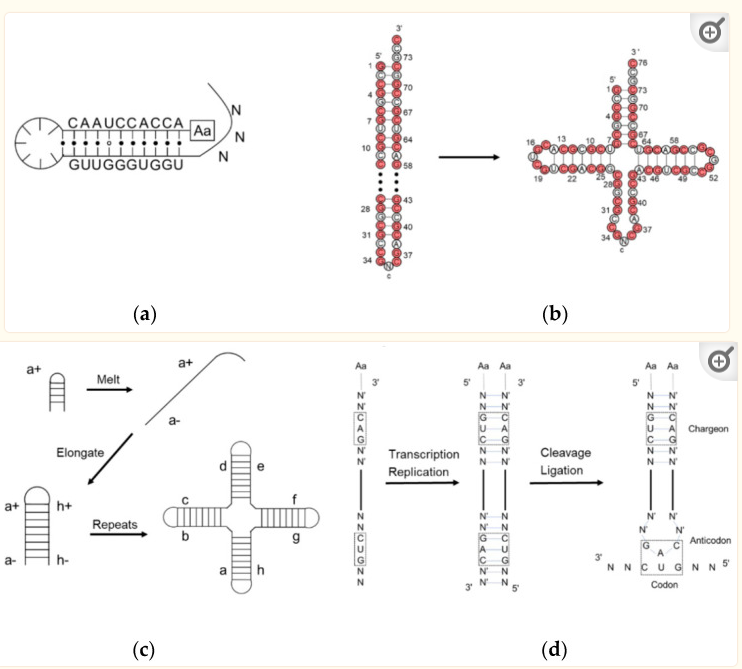
Abb. 19: Frühe Modelle der tRNA-Evolution. (a) Hopfield-Modell, das die Trinukleotid-Interaktion mit der Aminosäure am 5′-Terminus zeigt; (b) Winkler-Oswatitsch- und Eigen-Modell, das zeigt, wie tRNA aus RNY-Typ-Tripletts entstand; (c) Bloch-Modell, das zeigt, wie tRNA aus RNA-Selbstpriming und Selbsttemplierung entstand; (d) Moller-Modell, das zeigt, wie tRNA aus Strangreplikation und Ligation entstand.
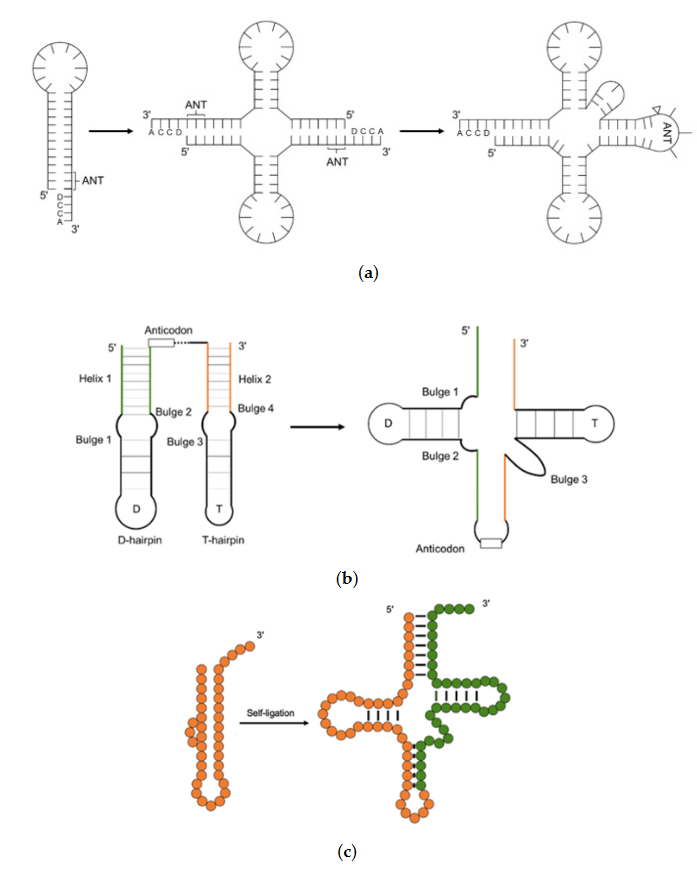
Abb. 20: Aktuelle Modelle der tRNA-Evolution. (a) Di-Giulio-Modell, das zeigt, wie tRNA aus Homodimer-Haarnadeln entstanden ist; (b) Tanaka-Modell, das zeigt, wie tRNA aus zwei unterschiedlichen Haarnadeln mit Ausbuchtungen entstanden ist; (c) Fox-Modell, das zeigt, wie tRNA aus zwei identischen Haarnadeln mit Ausbuchtungen durch Selbstligatur entstanden ist. ANT, Anticodon. D, Basebestimmer.
tRNA-Gene
Die Gene für die tRNA sind recht vielfältig. Das menschliche Genom hat z. B. über 400 tRNA-Gene zur Dekodierung von 61 Codons (Chan & Lowe 2016, Lowe & Eddy 1997), d. h. es gibt mehrere tRNA-Gene und tRNAs, die identische Anticodon-Sequenzen für eine bestimmte Aminosäure tragen. tRNAs mit gleichem Anticodon werden als „Isodekodierer“ bezeichnet, während tRNAs mit unterschiedlichem Anticodon, die aber mit denselben Aminosäuren geladen sind, „Isoakzeptoren“ genannt werden. Eine Überexpression und Mutation zellulärer und mitochondrialer tRNA steht in Zusammenhang mit einer Vielzahl menschlicher Krankheiten (Abbott et al. 2014, Brandon et al. 2005, Yarham et al. 2010, Pavon-Eternod et al. 2009, Glatz et al. 2011). Die DNA-Sequenz, die die tRNA kodiert, ist entweder durch Introns, also nicht codierende Abschnitte der DNS, getrennt, fragmentiert oder umgeordnet (Fujishima & Kanai 2014, Tocchini-Valentini et al. 2005). Aufspaltung und Fragmentierung in tRNA-Genen werden als späte Errungenschaften (Randau & Söll 2008) oder als Überbleibsel der frühen tRNA angesehen (Di Giulio 2006). Solche Fragmentierung von Genen durch Introns kommt in den meisten Genen der Eukaryoten vor und ist höchstwahrscheinlich das Ergebnis ihrer Entstehung – ein Prozess, den wir uns gesondert ansehen werden.
Archaeen weisen die einfachste Situation in Bezug auf den genomischen tRNA-Gehalt mit einer einheitlichen Anzahl von Genkopien auf, Bakterien eine mittlere Komplexität und Eukaryoten die höchste Komplexität (Novoa et al. 2012). Eukaryoten weisen nicht nur einen höheren tRNA-Gehalt als die anderen beiden Reiche auf, sondern auch eine hohe Variation der Genkopienzahl und diese Komplexität scheint auf Duplikationen von tRNA-Genen und Änderungen der Anticodon-Spezifität zurückzuführen zu sein.
Viele Arten haben im Laufe der Evolution bestimmte tRNAs verloren. So fehlen z. B. sowohl Säugetieren als auch Vögeln die gleichen 14 der möglichen 64 tRNA-Gene, aber andere Lebensformen enthalten diese tRNAs (Ou et al. 2020). Für die Übersetzung von Codons, für die eine exakt passende tRNA fehlt, greifen Organismen auf eine Strategie zurück, die als Wobbling bezeichnet wird (Abb. 21) und bei der unvollkommen passende tRNA/mRNA-Paare dennoch zu einer Übersetzung führen, obwohl diese Strategie auch die Neigung zu Übersetzungsfehlern erhöht (Ou et al. 2019). Die Gründe, warum tRNA-Gene im Laufe der Evolution verloren gegangen sind, werden nach wie vor diskutiert, könnten aber mit der Verbesserung der Resistenz gegen Virusinfektionen zusammenhängen (Ou et al. 2018).
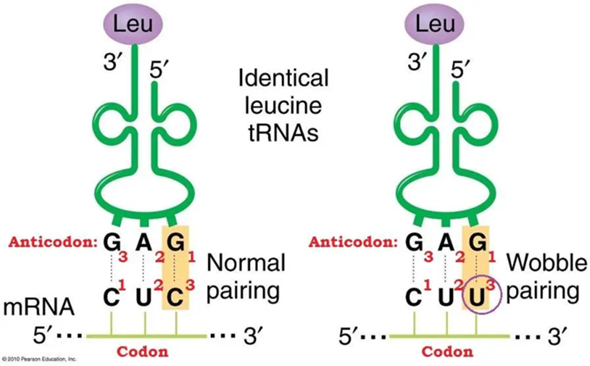
Abb. 21: Die wobble-Hypothese besagt, dass die 1. Anticodon-Base als Partner in der 3. Codon-Position auch Alternativen zulässt, also schwankt (engl.: to wobble), während Base 1 und 2 des Codons der mRNA normale Paarungen eingehen. Das Anticodon der aminosäurebeladenen tRNA hat in diesem Beispiel die Basenfolge GAG. Normal wäre die Paarung CUC als Codon. Die gleiche t-RNA erkennt aber auch CUU (rechts), wobei ein G und ein U paaren. Das G an der 1. Anticodonstelle lässt also Alternativen zum C zu. So dass beide Codons CUC und CUU, obwohl sie in der 3. Base variieren (wobbeln), von der gleichen tRNA (GAG) erkannt werden und zur gleichen Aminosäure (Leucin) führen.
Ribosomen
Die Hauptaufgabe eines Ribosoms ist die Translation während der Proteinbiosynthese. Ein Ribosom besteht aus zwei Untereinheiten: die kleine Untereinheit dient dem Ablesen und der Kontrolle der mRNA, die große Untereinheit sorgt für die Bildung der Proteine. Ribosomen bestehen neben den Proteinen auch aus rRNA (Munk 2009, Abb. 22).
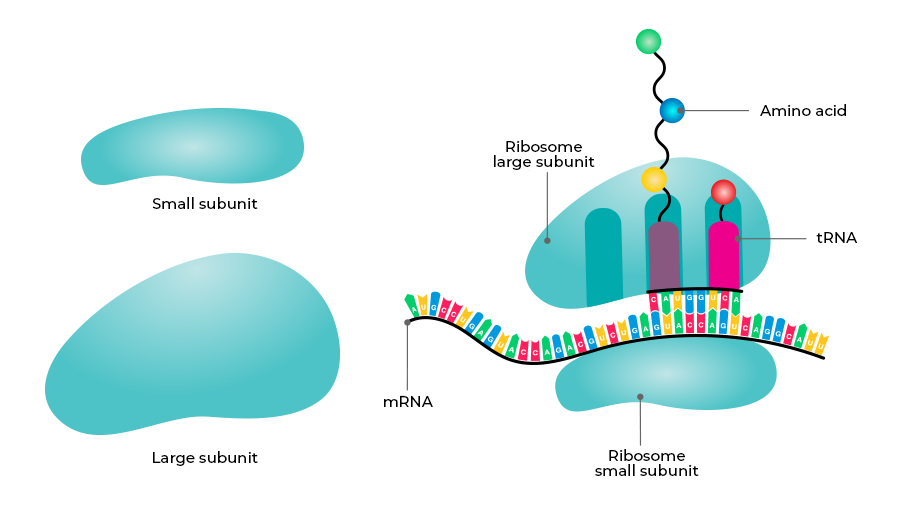
Abb. 22: Struktur des Ribosoms
Bei Eukaryoten und Prokaryoten sind die Ribosomen und ihre Untereinheiten unterschiedlich groß, sind sich aber untereinander recht ähnlich. Um sie vergleichen zu können, gibt es den sogenannten Sedimentationskoeffizienten. Er hat die Einheit Svedberg (S) und hängt von Masse und Form des Teilchens ab. Der Koeffizient misst dabei, wie schnell ein Teilchen in einer Zentrifuge absinken würde. Prokaryoten haben 70S Ribosomen. Ihre große 50S- Untereinheit (50S) besteht dabei aus 31 Proteinen und zwei verschiedenen rRNA Molekülen (23S und 5S). Die kleine 30S-Untereinheit (30S) besteht nur aus einem 16S-rRNA Molekül und 21 Proteinen. Eukaryoten haben 80S Ribosomen, deren große 60S-Untereinheit aus 49 Proteinen und drei RNA-Molekülen (28, 5,8 und 5S) besteht. Die kleine Untereinheit hat 33 Proteine und ein 18S-RNA-Molekül. Während prokaryotische Ribosomen zu 65% aus rRNA und 35% Proteinen bestehen, ist das Verhältnis bei eukaryotischen Ribosomen etwa 1 zu 1. Die Proteine der kleinen Untereinheit werden mit „s“ (englisch small ‚klein‘), die der großen Untereinheit mit „L“ (engl. large ‚groß‘) gekennzeichnet. Ihre Aminosäuresequenzen besitzen keine besonderen Gemeinsamkeiten, sind aber reich an positiv geladenen Aminosäuren wie L-Lysin oder L-Arginin. Dies erlaubt eine bessere Interaktion mit den negativ geladenen rRNAs. In Eukaryoten gibt es außer den freien cytoplasmatischen Ribosomen auch membrangebundene Ribosomen, die an die Membran des rauen Endoplasmatischen Retikulums (ER) gebunden sind (Kurland 1960, Wilson & Doudna Cate 2012, Yusupov et al. 2001, Yusupova et al. 2006, Lafontaine & Tollervey 2001, Wimberly et al. 2000, Schuwirth et al. 2005, Ben-Shem et al. 2011, Rabl et al. 2011, Klinge et al. 2011, Abb. 23).
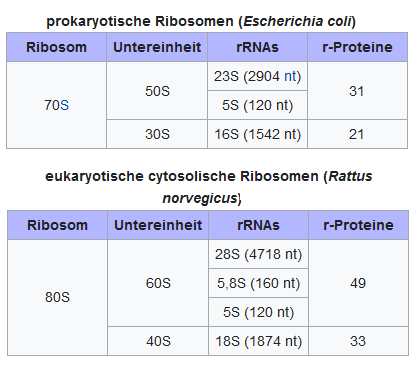
Abb. 23: Unterschiede zwischen prokaryotischen und eukaryotischen Ribosomen.
Kristallographische Arbeiten (Nissen et a. 2000, Mitra et al. 2005) haben gezeigt, dass es keine ribosomalen Proteine in der Nähe der Reaktionsstelle für die Polypeptidsynthese gibt. Dies deutet darauf hin, dass die Proteinkomponenten der Ribosomen nicht direkt an der Katalyse der Peptidbindungsbildung beteiligt sind, sondern dass diese Proteine vielmehr als Gerüst fungieren, das die Fähigkeit der rRNA zur Proteinsynthese verbessern kann.
Das Ribosom könnte zunächst als Protoribosom (Dance 2023, Agmon 2024, Abb. 24), das möglicherweise ein Peptidyltransferasezentrum (PTC) enthält (Nissen et al. 2000, Noller et al. 1992, Gregory & Dahlberg 2004, Codispoti et al. 2024) in einer RNA-Welt entstanden sein und als selbstreplizierender Komplex erscheinen, der erst später, als Aminosäuren auftauchten, die Fähigkeit zur Proteinsynthese entwickelte (Nissen 2012). Studien legen nahe, dass präbiotische Ribosomen, die ausschließlich aus rRNA bestehen, die Fähigkeit zur Synthese von Peptidbindungen entwickelt haben könnten (Nomura et al. 1969, Dabbs 1986, Noller et al. 1992, Krupkin et al. 2011, Bose et al. 2022, Codispoti et al. 2024).
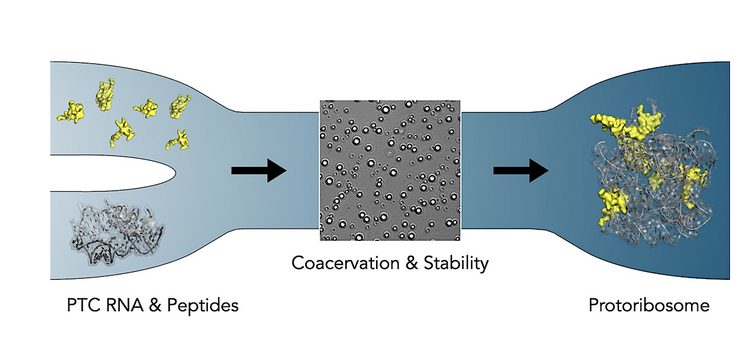
Abb. 24: Das Ribosom beherbergt aufgrund seiner außergewöhnlichen Erhaltung ein bemerkenswertes molekulares Fossil, das als Protoribosom bekannt ist. Es umgibt das Peptidyltransferasezentrum (PTC), das für die Bildung von Peptidbindungen verantwortlich ist.
Es wird angenommen, dass die Region des Peptidyltransferase-Zentrums (PTC) in der modernen großen ribosomalen Untereinheit ein Überbleibsel eines solchen präbiotischen, nicht kodierten Protoribosoms enthält, das sich selbst aus zufälligen RNA-Ketten zusammengesetzt, die Bildung von Peptidbindungen zwischen beliebigen Aminosäuren katalysiert und kurze Peptide produziert hätte. Kürzlich haben drei Forschungsgruppen experimentell nachgewiesen, dass verschiedene Konstrukte von Protoribosom-Analoga die Fähigkeit besitzen, sich spontan zu falten und die Bildung von Peptidbindungen und kurzen Peptiden zu katalysieren (Xu & Wang 2021, Bose et al. 2022, Kawabata et al. 2022).
Darüber hinaus gibt es starke Hinweise darauf, dass urtümliche Ribosomen selbstreplizierende Komplexe waren, bei denen die rRNA in den Ribosomen informatorische, strukturelle und katalytische Aufgaben hatte, da sie für tRNAs und Proteine kodiert haben könnte, die für die ribosomale Selbstreplikation benötigt werden (Root-Bernstein & Root-Bernstein 2015, Yarus 2002, Forterre & Krupovic 2012).
Als Aminosäuren unter präbiotischen Bedingungen allmählich in der RNA-Welt auftauchten (Caetano-Anolles & Seufferheld 2013, Saladino et al. 2012), würden ihre Wechselwirkungen mit katalytischer RNA sowohl die Reichweite als auch die Effizienz der Funktion katalytischer RNA-Moleküle erhöhen (Noller 2012). Die treibende Kraft für die Evolution des Ribosoms von einer uralten selbstreplizierenden Maschine zu seiner heutigen Form als Translationsmaschine könnte also der Selektionsdruck gewesen sein, Proteine in die selbstreplizierenden Mechanismen des Ribosoms einzubauen, um seine Fähigkeit zur Selbstreplikation zu erhöhen (Root-Bernstein & Root-Bernstein 2015, Fox 2010, 2016).
Die Entstehung des Genoms ermöglichte ein komplexer werden des Lebens, inklusive verschiedener Stoffwechselwege. Doch wie wahrscheinlich ist es überhaupt, dass das Leben entstehen konnte. Ist dieser blinde Zufall nicht doch zu unwahrscheinlich? Dies werden wir in der nächsten Episode behandeln.
Literatur
Abbott J.A., Francklyn C.S., Robey-Bond S.M. (2014): Transfer RNA and human disease. Front. Genet. 5:158.
Agmon I. (2024): On the Re-Creation of Protoribosome Analogues in the Lab. Int J Mol Sci. 25(9): 4960.
Barrell, B., Bankier, A. Drouin, J. (1979): A different genetic code in human mitochondria. Nature 282, 189–194
Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M (2011): The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 334 (6062): 1524–9.
Bloch D.P., McArthur B., Mirrop S. (1985): tRNA-rRNA sequence homologies: Evidence for an ancient modular format shared by tRNAs and rRNAs. Biosystems 17:209–225.
Bose T, Fridkin G, Davidovich C, Krupkin M, Dinger N, Falkovich AH, Peleg Y, Agmon I, Bashan A, Yonat A (2022): Origin of life: protoribosome forms peptide bonds and links RNA and protein dominated worlds. Nucleic Acids Res. 50 (4): 1815–1828.
Brandon M.C., Lott M., Nguyen K.C., Spolim S., Navathe S.B., Baldi P., Wallace D.C. (2005): MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 33((Suppl. S1)):D611–D613.
Burton Z.F. (2020): The 3-minihelix tRNA evolution theorem. J. Mol. Evol. 88:234–242.
Caetano-Anollés G, Seufferheld MJ (2013): The coevolutionary roots of biochemistry and cellular organization challenge the RNA world paradigm. Journal of Molecular Microbiology and Biotechnology. 23 (1–2): 152–77.
Černý, J., Bolfíková, B. Č., Valdés, J. J., Grubhoffer, L., Růžek, D. (2014): Evolution of tertiary structure of viral RNA dependent polymerases. PLOS ONE 9:e96070.
Chan P.P., Lowe T.M. (2016): GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44:D184–D189.
Codispoti, S., Yamaguchi, T., Makarov, M., Giacobelli, V. G., Mašek, M., Kolář, M. H., Rocha, A. C. S., Fujishima, K., Zanchetta, G., Hlouchová, K. (2024): The interplay between peptides and RNA is critical for protoribosome compartmentalization and stability, Nucleic Acids Research, gkae823
Copley SD, Smith E, Morowitz HJ (2005): A mechanism for the association of amino acids with their codons and the origin of the genetic code. Proc Natl Acad Sci U S A 102(12):4442-7.
Crick, F. (1967): Origin of the genetic code. Nature 213:119. doi: 10.1038/213119d0.
Crick, F., Barnett, L., Brenner, S., Watts-Tobin, R. J. (1961): General nature of the genetic code for proteins, Nature 192, S. 1227–1232
Dabbs ER (1986): Mutant studies on the prokaryotic ribosome. New York: Springer-Verlag.
Dance, A. (2023): How did life begin? One key ingredient is coming into view – A Nobel-prizewinning scientist’s team has taken a big step forward in its quest to reconstruct an early-Earth RNA capable of building proteins. Nature. 615 (7950): 22–25.
de Farias ST, Dos Santos Junior AP, Rêgo TG, José MV (2017): Origin and Evolution of RNA-Dependent RNA Polymerase. Front Genet 8:125.
De Farias, S. T., Rêgo, T. G., José, M. V. (2016): A proposal of the proteome before the last universal common ancestor. Int. J. Astrobiol. 15, 27–31.
Demongeot J., Moreira A. (2007): A possible circular RNA at the origin of life. J. Theor. Biol. 249:314–324.
Demongeot J., Seligmann H. (2019): Spontaneous evolution of circular codes in theoretical minimal RNA rings. Gene 705:95–102.
Di Giulio M. (1992): On the origin of the transfer RNA molecule. J. Theor. Biol. 159:199–214
Di Giulio M. (2004): The origin of the tRNA molecule: Implications for the origin of protein synthesis. J. Theor. Biol. 226:89–93.
Di Giulio M. (2006): The non-monophyletic origin of the tRNA molecule and the origin of genes only after the evolutionary stage of 1the last universal common ancestor (LUCA) J. Theor. Biol. 240:343–352.
Di Giulio M. (2020): An RNA ring was not the progenitor of the tRNA molecule. J. Mol. Evol. 88:228–233.
Doig A.J. (2017): Frozen, but no accident—Why the 20 standard amino acids were selected. FEBS J. 284:1296–1305.
Dunnill P. (1966): Triplet nucleotide-amino-acid pairing; a stereochemical basis for the division between protein and non-protein amino-acids. Nature. 210:1265–1267.
Eigen M., Winkler-Oswatitsch R. (1981): Transfer-RNA, an early gene? Naturwissenschaften 68:282–292.
Ellington AD, Khrapov M, Shaw CA (2000): The scene of a frozen accident. RNA 6:485– 498.
Forterre P, Krupovic M (2012): The Origin of Virions and Virocells: The Escape Hypothesis Revisited. Viruses: Essential Agents of Life. pp. 43–60.
Fox GE (2010): Origin and Evolution of the Ribosome. Cold Spring Harb Perspect Biol. 2 (9): a003483.
Fox GE (2016): Origins and early evolution of the ribosome. In Hernández G, Jagus R (eds.). Evolution of the Protein Synthesis Machinery and Its Regulation. Switzerland: Springer, Cham. S. 31–60.
Freeland SJ, Hurst LD (1998): The genetic code is one in a million. J Mol Evol. 47(3):238-48.
Fujishima K., Kanai A. (2014): tRNA gene diversity in the three domains of life. Front. Genet. 5:142.
Gamow, G. (1954a): Possible relation between DNA and protein structures, Nature 173, S. 318
Gamow, G. (1954b): Possible mathematical relation between Deoxyribonucleic Acid and Proteins, Kong. Danske Vid. Selsk., Biolog. Meddelelser, Band 22, Nr. 3
Gilbert, W. (1986): The RNA world. Nature 319:618.
Glatz C., D’Aco K., Smith S., Sondheimer N. (2011): Mutation in the mitochondrial tRNA(Val) causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes. Mitochondrion. 11:615–619.
Gregory, S.T.; Dahlberg, A.E. (2004): Peptide bond formation is all about proximity. Nat. Struct. Mol. Biol. 11, 586–587.
Guo X, Su M. (2022): The Origin of Translation: Bridging the Nucleotides and Peptides. Int J Mol Sci. 24(1):197.
Harvey S.C., McCammon J.A.(1981): Intramolecular flexibility in phenylalanine transfer RNA. Nature. 294:286–287.
Hoagland M.B., Stephenson M.L., Scott J.F., I Hecht L., Zamecnik P.C.(1958): A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem 231:241–257.
Hopfield J.J. (1978): Origin of the genetic code: A testable hypothesis based on tRNA structure, sequence, and kinetic proofreading. Proc. Natl. Acad. Sci. USA. 75:4334–4338.
Horning, D. P., Joyce, G. F. (2017): Amplification of RNA by an RNA polymerase ribozyme. Proc. Natl. Acad. Sci. U.S.A. 113, 9786–9791.
Hou Y.M., Schimmel P. (1988): A simple structural feature is a major determinant of the identity of a transfer RNA. Nature 333:140–145.
Hou Y.M., Schimmel P. (1989): Evidence that a major determinant for the identity of a transfer RNA is conserved in evolution. Biochemistry 28:6800–6804.
Jácome, R., Becerra, A., León, S. P., Lazcano, A. (2015): Structural analysis of monomeric RNA dependent polymerases: evolutionary and therapeutic implications. PLOS ONE 10:e0139001.
Johnson D.B., Wang L. (2010): Imprints of the genetic code in the ribosome. Proc. Natl. Acad. Sci. USA. 107:8298–8303.
Jovine L., Djordjevic S., Rhodes D. (2000): The crystal structure of yeast phenylalanine tRNA at 2.0 A resolution: Cleavage by Mg2+ in 15-year old crystals. J. Mol. Biol. 301:401–414.
Judson, H. (1996): The eighth day of creation, Cold Spring Harbor Press
Kawabata, M.; Kawashima, K.; Mutsuro-Aoki, H.; Ando, T.; Umehara, T.; Tamura, K. (2022): Peptide Bond Formation between Aminoacyl-Minihelices by a Scaffold Derived from the Peptidyl Transferase Center. Life 12, 573.
Kim, Y. E., Higgs, P. G. (2016): Co-operation between polymerases and nucleotide synthetases in the RNA world. PLOS Comput. Biol. 12:e1005161.
Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N (2011): Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 334 (6058): 941–8.
Knight RD, Landweber LF. (2000): Guilt by association: the arginine case revisited. RNA 6(4):499-510.
Koonin EV (2017): Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code. Life (Basel) 7(2):22.
Koonin EV, Novozhilov AS (2009): Origin and evolution of the genetic code: the universal enigma. IUBMB Life. 61(2):99-111.
Krupkin M, Matzov D, Tang H, Metz M, Kalaora R, Belousoff MJ, Zimmerman E, Bashan A, Yonath A (2011): A vestige of a prebiotic bonding machine is functioning within the contemporary ribosome. Phil. Trans. R. Soc. B. 366 (1580): 2972–8.
Kühnlein, A.; Lanzmich, S. A.; Brun, D. ( 2021): tRNA sequences can assemble into a replicator. eLife. 10.
Kurland CG (1960): Molecular characterization of ribonucleic acid from Escherichia coli ribosomes. Journal of Molecular Biology. 2 (2): 83–91.
Kwapisz M, Beckouët F, Thuriaux P. (2008): Early evolution of eukaryotic DNA-dependent RNA polymerases. Trends Genet 24(5):211-5.
Lafontaine, D.; Tollervey, D. (2001): The function and synthesis of ribosomes. Nat Rev Mol Cell Biol. 2 (7): 514–520.
Lowe T.M., Eddy S.R. (1997): tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
Ludwig, M. (2021): Solving the Chicken-and-the-Egg Problem – „A Step Closer to the Reconstruction of the Origin of Life“. SciTechDaily.
Masta S.E., Boore J.L. (2008): Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol. Biol. Evol. 25:949–959.
Maizels N., Weiner A.M. (1994): Phylogeny from function: Evidence from the molecular fossil record that tRNA originated in replication, not translation. Proc. Natl. Acad. Sci. USA. 91:6729–6734.
Maizels, N.; Weiner, A. M. (1999): The Genomic Tag Hypothesis – What Molecular Fossils Tell Us about the Evolution of tRNA. The RNA World (2nd ed.). Cold Spring Harbor Laboratory Press.
Mitra K, Schaffitzel C, Shaikh T, Tama F, Jenni S, Brooks CL, Ban N, Frank J (2005): Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature. 438 (7066): 318–24.
Möller W., Janssen G.M. (1992): Statistical evidence for remnants of the primordial code in the acceptor stem of prokaryotic transfer RNA. J. Mol. Evol 34:471–477.
Müller, U. F. (2006): Re-creating an RNA world. Cell Mol. Life Sci. 63, 1278–1293.
Munk, K. (2009, Hrsg.): Taschenlehrbuch Biologie: Genetik. Stuttgart: Thieme Verlag
Naganuma M., Sekine S.-I., Chong Y.E., Guo M., Yang X.-L., Gamper H., Hou Y.-M., Schimmel P., Yokoyama S. (2014): The selective tRNA aminoacylation mechanism based on a single G•U pair. Nature 510:507–611.
Nagaswamy U., Fox G.E. (2003): RNA ligation and the origin of tRNA. Orig. Life Evol. Biosph. 33:199–209.
Nasir, A., Caetano-Anollés, G. A. (2015): A phylogenomic data-driven exploration of viral origins and evolution. Sci. Adv. 1:e1500527.
Neukamm, M. (2021): Evolution: Die Entstehung des genetischen Codes. Zufall oder perfektes Design – was sagt die Wissenschaft? https://www.ag-evolutionsbiologie.de/pdf/2021/der-genetische-code-entstehung-zufall-oder-perfektes-design.pdf
Neukamm, M., Kaiser, P. M. (2014): Chemische Evolution und evolutionäre Bioinformatik. In: Neukamm, M. (Hrsg): Darwin heute Evolution als Leitbild in den modernen Wissenschaften.Darmstadt: WBG
Nirenberg M., Leder P., Bernfield M., Brimacombe R., Trupin J., Rottman F., O’Neal C. (1965): RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proc. Natl. Acad. Sci. USA. 53:1161–1168.
Nissen P, Hansen J, Ban N, Moore PB, Steitz TA (2000): The structural basis of ribosome activity in peptide bond synthesis. Science. 289 (5481): 920–30.
Noller HF (2012): Evolution of protein synthesis from an RNA world. Cold Spring Harbor Perspectives in Biology. 4 (4): a003681.
Noller, H.F.; Hoffarth, V.; Zimniak, L. (1992): Unusual Resistance of Peptidyl Transferase to Protein Extraction Procedures. Science 256, 1416–1419.
Nomura M, Mizushima S, Ozaki M, Traub P, Lowry CV (1969): Structure and function of ribosomes and their molecular components. Cold Spring Harbor Symposia on Quantitative Biology. 34: 49–61.
Novoa EM, Pavon-Eternod M, Pan T, Ribas de Pouplana L (2012): A role for tRNA modifications in genome structure and codon usage. Cell. 149 (1): 202–213.
Novozhilov AS, Wolf YI, Koonin EV (2007): Evolution of the genetic code: partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol Direct. 2:24.
Ohtsuki T., Kawai G., Watanabe K. (2002): The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNA(Ser)(UCU) having a short T arm and lacking the entire D arm. FEBS Lett. 514:37–43.
Ohtsuki T., Watanabe Y. (2007): T-armless tRNAs and elongated elongation factor Tu. IUBMB Life. 59:68–75.
Ou X, Cao J, Cheng A, Peppelenbosch MP, Pan Q (2019): Errors in translational decoding: tRNA wobbling or misincorporation?. PLOS Genetics. 15 (3): 2979–2986.
Ou X, Peng W, Yang Z, Cao J, Wang M, Peppelenbosch MP, Pan Q, Cheng A (2020): Evolutionarily missing and conserved tRNA genes in human and avian. Infect. Genet. Evol. 85: 104460.
Ou X, Wang M, Mao S, Cao J, Cheng A, Zhu D, Chen S, Jia R, Liu M, Yang Q, Wu Y, Zhao X, Zhang S, Liu Y, Yu Y, Zhang L, Chen X, Peppelenbosch MP, Pan Q (2018): Incompatible Translation Drives a Convergent Evolution and Viral Attenuation During the Development of Live Attenuated Vaccine. Front. Cell. Infect. Microbiol. 8: 249.
Pak D., Root-Bernstein R., Burton Z.F. (2017): tRNA structure and evolution and standardization to the three nucleotide genetic code. Transcription. 8:205–219.
Pavon-Eternod M., Gomes S., Geslain R., Dai Q., Rosner M.R., Pan T. (2009): tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 37:7268–7280.
Pelc S.R. (1965): Correlation between coding-triplets and amino-acids. Nature 207:597–599.
Pelc S.R., Welton M.G. (1966): Stereochemical relationship between coding triplets and amino-acids. Nature 209:868–870.
Petrov A.S., Gulen B., Norris A.M., Kovacs N.A., et al. (2015): History of the ribosome and the origin of translation. Proc. Natl. Acad. Sci. USA. 112:15396–15401.
Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N (2011): Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 331 (6018): 730–6.
Randau L., Söll D. (2008): Transfer RNA genes in pieces. EMBO Rep. 9:623–628.
Rodin A.S., Szathmáry E., Rodin S.N. (2011): On origin of genetic code and tRNA before translation. Biol. Direct. 6:14.
Root-Bernstein M, Root-Bernstein R (2015): The ribosome as a missing link in the evolution of life. Journal of Theoretical Biology. 367: 130–158.
Saladino R, Botta G, Pino S, Costanzo G, Di Mauro E (2012): Genetics first or metabolism first? The formamide clue. Chemical Society Reviews. 41 (16): 5526–65.
Santos MA, Moura G, Massey SE, Tuite MF (2004): Driving change: the evolution of alternative genetic codes. Trends Genet 20(2):95-102.
Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH (2005): Structures of the bacterial ribosome at 3.5 A resolution. Science. 310 (5749): 827–34.
Sella G, Ardell DH (2006): The coevolution of genes and genetic codes: Crick’s frozen accident revisited. J Mol Evol 63(3):297-313.
Shechner, D. M., Grant, R. A., Bagby, S. C., Koldobskaya, Y., Piccirilli, J. A., Bartel, D. P. (2009): Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science 326, 1271–1275.
Söll D, RajBhandary UL (2006): The genetic code – thawing the ‚frozen accident‘. J Biosci. 31(4):459-63.
Sun F.J., Caetano-Anollés G. (2008): The origin and evolution of tRNA inferred from phylogenetic analysis of structure. J. Mol. Evol. 66:21–35.
Szathmáry E. (1993): Coding coenzyme handles: A hypothesis for the origin of the genetic code. Proc Natl Acad Sci USA 90:9916–9920.
Szathmáry E. (1999): The origin of the genetic code: Amino acids as cofactors in an RNA world. Trends Genet 15:223–229.
Tagami, S., Attwater, J., Holliger, P. (2017): Simple peptides derived from the ribosomal core potentiate RNA polymerase ribozyme function. Nat. Chem. 9, 325–332.
Tanaka T., Kikuchi Y. (2001): Origin of the cloverleaf shape of transfer RNA—The double-hairpin model: Implication for the role of tRNA intron and the long extra loop. Viva Origino. 29:134–142.
Taylor FJ, Coates D. (1989): The code within the codons. Biosystems. 22(3):177-87.
Tocchini-Valentini G.D., Fruscoloni P., Tocchini-Valentini G.P. (2005): Coevolution of tRNA intron motifs and tRNA endonuclease architecture in Archaea. Proc. Natl. Acad. Sci. USA. 102:15418–15422.
Trifonov E.N. (2000): Consensus temporal order of amino acids and evolution of the triplet code. Gene. 261:139–151.
Trifonov E.N. (2004): The triplet code from first principles. J. Biomol. Struct. Dyn. 22:1–11.
Weber A.L., Lacey J.C., Jr. (1978): Genetic code correlations: Amino acids and their anticodon nucleotides. J. Mol. Evol. 11:199–210.
Weberndorfer G, Hofacker IL, Stadler PF (2003): On the evolution of primitive genetic codes. Orig Life Evol Biosph 33(4-5):491-514.
Weitze, M.-D. (2006): Zwischen Evolution und Engineering – Der genetische Code im Wandel. Biologie in unserer Zeit 1, 18–24.
Widmann J., Di Giulio M., Yarus M., Knight R. (2005): tRNA creation by hairpin duplication. J. Mol. Evol. 61:524–530.
Wilson DN, Doudna Cate JH (2012): The structure and function of the eukaryotic ribosome. Cold Spring Harbor Perspectives in Biology. 4 (5): a011536.
Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V (2000): Structure of the 30S ribosomal subunit. Nature. 407 (6802): 327–39.
Wnętrzak, M., Blazej, P., Mackiewicz, D., Mackiewicz, P. (2018): The optimality of the standard genetic code assessed by an eight-objective evolutionary algorithm. BMC Evolutionary Biology. 18.
Woese C.R. (1969): The biological significance of the genetic code. In: Hahn F.E., editor. Progress in Molecular and Subcellular Biology. Springer; Berlin/Heidelberg, Germany, S. 5–46.
Wu HL, Bagby S, van den Elsen JM (2005): Evolution of the genetic triplet code via two types of doublet codons. J Mol Evol. 61(1):54-64.
Wu L.-F., Liu Z., Roberts S.J., Su M., Szostak J.W., Sutherland J.D. (2022): Template-free assembly of functional RNAs by loop-closing ligation. J. Am. Chem. Soc. 144:13920–13927.
Xu, D.; Wang, Y. (2021): Protein-free ribosomal RNA scaffolds can assemble poly-lysine oligos from charged tRNA fragments. Biochem. Biophys. Res. Commun. 544, 81–85.
Yarham J.W., Elson J.L., Blakely E.L., McFarland R., Taylor R.W. (2010): Mitochondrial tRNA mutations and disease. Wiley Interdiscip. Rev. RNA. 1:304–324.
Yarus M. (1998): Amino acids as RNA ligands: A Direct-RNA-Template theory for the code’s origin. J Mol Evol 47:109–117.
Yarus M (2002): Primordial genetics: phenotype of the ribocyte. Annual Review of Genetics. 36: 125–51.
Yarus M, Widmann JJ, Knight R (2009): RNA-amino acid binding: a stereochemical era for the genetic code. J Mol Evol. 69(5):406-29.
Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF (2001): Crystal structure of the ribosome at 5.5 A resolution. Science. 292 (5518): 883–96.
Yusupova G, Jenner L, Rees B, Moras D, Yusupov M (2006): Structural basis for messenger RNA movement on the ribosome. Nature. 444 (7117): 391–4.
Zong, J., Yao, X., Yin, J., Zhang, D., Ma, H. (2009): Evolution of the RNA-dependent RNA polymerase (RdRP) genes: duplications and possible losses before and after the divergence of major eukaryotic groups. Gene 447, 29–39.