Wir haben verschiedene Zellorganellen schon kennengelernt, allen voran Mitochondrien und Chloroplasten bei Zellatmung und Photosynthese, den Zellkern bei der DNA und die Grundlagen der Biomembranen.
In diesem Beitrag wenden wir uns weiteren Zellorganellen zu, vornehmlich dem endoplasmatischen Reticulum und dem Golgi-Apparat.
Endoplasmatisches Reticulum (ER) und Golgi-Apparat (GA)
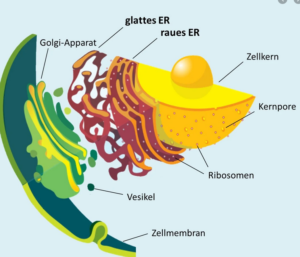
Abb. 1: Endoplasmatisches Reticulum
Das endoplasmatische Retikulum liegt in zwei Formen vor, dem glatten ER (SER) und dem rauen ER (RER). Auf dem rauen ER befinden sich die Ribosomen, kleine Zellorganellen, die bei der Produktion von Proteinen eine wichtige Rolle spielen.
Das endoplasmatische Retikulum erfüllt mehrere wichtige Aufgaben. Grundsätzlich ist es für die Signalübertragung innerhalb der Zelle verantwortlich. Diese Signalübertragung kann man sich als eine Aufnahme von Calcium-Ionen vorstellen. Das ist insbesondere bei Muskelzellen und Nervenzellen sehr wichtig.
Das glatte ER ist vor allem bei Stoffwechselvorgängen wichtig. Enzyme, die sich innerhalb des glatten ER befinden, können Lipide herstellen.
Weil sich am rauen ER die Ribosomen befinden, ist dieses vor allem bei der Herstellung von Proteinen beteiligt. Eine weitere Hauptfunktion des rauen ER ist die Bildung einer Kernmembran.
Das sogenannte Sarkoplasmatische Retikulum (SR) ist eine spezielle Form des endoplasmatischen Retikulums. Es befindet sich insbesondere in der glatten und der quergestreiften Muskulatur, besonders in den Muskelfasern. In der Nähe des SR befinden sich auch viele Mitochondrien.
Die Hauptaufgabe des SR ist die Speicherung von Calcium-Ionen. Wenn ein Nervensignal an die Muskeln geliefert wird, schüttet das SR die Calcium-Ionen in das Plasma der Muskelzellen aus. Dadurch sind die Calcium-Ionen in der Lage, ein Zusammenziehen des Muskels (Kontraktion) einzuleiten. Somit ist das SR ein wichtiger Vermittler, um jede deiner Bewegungen ausführen zu können.
Ein weiteres wichtiges internes Membransystem stellt der Golgi-Apparat dar (siehe Abb. 1). Eine seiner Aufgaben ist es, Proteine zu verarbeiten und umzuwandeln. Die Proteine kommen dabei vor allem vom endoplasmatischen Retikulum (ER). Außerdem ist er in der Lage, diese Proteine in Vesikeln zu binden. Diese können die Proteine zu verschiedensten Bereichen innerhalb der Zelle transportieren.
Der Golgi-Apparat besteht aus mehreren gestapelten Membran-Zisternen, die auch als Dictyosomen bezeichnet werden. Die Gesamtheit der Dictyosomen innerhalb der Zelle wird als Golgi-Apparat bezeichnet.
Um miteinander zu kommunizieren und Stoffe auszutauschen, sind die Dictyosomen über Kanäle miteinander verbunden.
Die Golgi-Stapel sind asymmetrisch angeordnet. Man unterscheidet einen cis-, einen medialen-, einen trans-Golgi-Bereich und ein trans-Golgi-Netzwerk (TGN). Jedes dieser Subkompartimente besitzt zwar spezifische Marker, doch die Grenzen sind fließend.
Proteinmodifikation und -transport
Das glatte ER kommt vermehrt in Zellen vor, die sich auf die Lipidsynthese spezialisiert haben oder in steroidhormonproduzierenden Zellen. In den Membranen des ausgedehnten glatten ERs muss genügend Platz für die Enzyme der Cholesterin-synthese und der Hormonproduktion sein. In den Membranen sind Enzyme lokalisiert, die die Lipid-anteile der Lipoproteine synthetisieren. Lipoproteine sind notwendig, um Lipide über die Blutbahn zu anderen Zielzellen zu transportieren. Andere Enzyme sind an der Beseitigung fettlöslicher Medikamente und Giftstoffe beteiligt.
Wie funktionierten die Proteinherstellung und der Transport im ER?
Was mit dem Protein geschieht, hängt ab vom Ort seiner Synthese. Wir erinnern uns: Proteine, bzw. die Polypeptidkette, werden mittels Translation an den Ribosomen synthetisiert.
Enthält die wachsende Polypeptidkette ein ER-Signalpeptid, wird sie noch während der Synthese, d. h. co-translational, zusammen mit den Ribosomen zur Membran des ER dirigiert. Im Gegensatz dazu gelangen Proteine, die an freien Ribosomen im Zellplasma synthetisiert werden, erst nach vollendeter Synthese, post-translational, zum Zielkompartiment. Über diesen Transportvorgang ist bislang wenig bekannt. Es wird angenommen, dass sog. Hsp70-Proteine an diesem Prozess beteiligt sind.
Beim Transport eines Proteins mit ER-Signalpeptid zur ER-Membran sind vor allem zwei zelluläre Komponenten notwendig: ein Signalerkennungspartikel (SRP, signal recognition particle) und der entsprechende Rezeptor (Abb. 2).
![]()
Abb. 2: Transport von Proteinen an das raue ER
Zuerst bindet das SRP an das Signalpeptid sobald es am Ribosom entstanden ist. Die Proteinsynthese stoppt und das SRP dirigiert die Polypeptidkette zusammen mit dem Ribosom zur ER-Membran. Der Komplex aus SRP, mRNA, Ribosom und Polypeptidkette bindet an den SRP-Rezeptor, ein Membranprotein, das auf der Zellplasma-Seite der ER-Membran lokalisiert ist und Bindungsstellen für GDP/GTP und SRP besitzt. GTP ist mit dem ATP verwandt. Bindet das SRP an den SRP-Rezeptor, wird aus dem GTP GDP+P. Durch diese Spaltung des GTP wird das SRP wieder freigesetzt.
Der Transport der wachsenden Polypeptidkette durch die Membran hindurch erfolgt durch den Translokationsapparat. Bindet ein Ribosom an den Translokationsapparat, bildet dieser eine Pore, die sich nach erfolgter Proteinsynthese und Ablösen des Ribosoms schließt. Die zentrale Rolle im Translokationsapparat spielt der Sec61-Komplex: Er wird bei der Signalerkennung der Polypeptidkette benötigt, wenn diese in den Translokationsapparat eindringt, ist verantwortlich für die enge Bindung der Ribosomen und schafft mit seinen drei verschiedenen Proteinen die entsprechende Membranumgebung für die Translokation der Polypeptidkette. Das Signalpeptid verbleibt im Translokationsapparat, während die wachsende Polypeptidkette kontinuierlich in Form einer Schleife durch die ER-Membran „hindurchsynthetisiert“ wird, bis sich das Protein vollständig im Lumen des ERs befindet, wo Chaperone die Faltung der Proteine unterstützen und überwachen. Chaperone sind Proteine, die neu synthetisierte Proteine bei der Faltung unterstützen. Als letztes wird das Signalpeptid durch das Enzym Signalpeptidase abgespalten. Dazu ist eine zusätzliche Erkennungssequenz erforderlich, die dem Signalpeptid folgt.
Die Glykosylierung ist eine der wichtigsten Funktionen des ERs (Abb. 3).
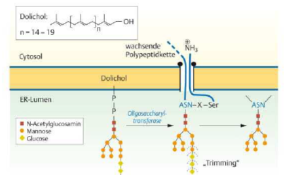
Abb. 3: Glykolysierung von Proteinen
Die meisten Proteine, die das ER verlassen und weiter zu Golgi-Apparat, Lysosomen, Cytoplasmamembran oder Umgebung der Zelle transportiert werden, sind Glykoproteine. Glykoproteine sind eine Verbindung von Proteinen mit Kohlenhydraten. Die Verbindung mit Zuckermolekülen an Proteinen verläuft im ER mittels des Enzyms Oligosaccharyl-Transferase. Dieses Enzym überträgt einen kompletten Oligosaccharidkomplex auf die NH2-Gruppe der Seitenkette eines Asparaginsäurerests. Hierfür gibt es spezielle Erkennungssequenzen aus drei Aminosäuren, an denen die Enzyme die Zuckermoleküle anheften können (Asp-X-Ser oder Asp-X-Thr, wobei X jede Aminosäure außer Prolin sein kann). Diese Übertragung wird auch als N-oder Asparagin-gekoppelte Glykosylierung bezeichnet und verläuft co-translational, also parallel zur Proteinsynthese. Durchweitere Modifikationen des Oligosaccharidkomplexes entsteht eine Vielfalt an Glykoproteinen. Bei den meisten Proteinen werden zunächst drei Glucosereste und eine Mannosegruppe entfernt. Dieses sogenannte Trimming beginnt im ER und wird im Golgi-Apparat fortgesetzt.
Oligosaccharide üben am Protein verschiedene Funktionen aus: Sie können das Protein vor Abbau schützen, das Protein im ER festhalten bis es korrekt gefaltet ist oder als Transportsignal dafür sorgen, dass das Protein in Vesikel verpackt und zum entsprechenden Kompartiment transportiert wird. Erscheinen Oligosaccharide auf der Zelloberfläche, so sind sie Teil der Glykokalyx und können an der Zell-Zell-Erkennung beteiligt sein.
Die Lipidsynthese findet an den Membranen des glatten ERs von Tier- und Pilzzellen statt. Sie betrifft Lipide, die zum Membranaufbau benötigt werden (Phospholipide, Cholesterin), Reservelipide (Triacylglycerol) und andere lipophile Verbindungen wie Steroide.
Die Phospholipidsynthese findet fast ausschließlich an der Zellplasma-Seite des ERs statt, da alle notwendigen Enzyme in der ER-Membran lokalisiert sind und ihre aktiven Zentren zum Zellplasma hin ausgerichtet haben. Damit sich die gesamte Lipiddoppelmembran vergrößern kann und das Wachstum nicht auf ein Membranblatt beschränkt bleibt, sind Phospholipid-Translokasen („Flippasen“) notwendig, die Phospholipidmoleküle auch zur luminalen Seite des ERs bringen. Diese Flippasen sind spezifisch für die entsprechende Kopfgruppe des Phospholipids.
Zwischen ER und Cytoplasmamembran, Golgi-Apparat, Lysosomen und Endosomen findet der Transport von Proteinen und Lipiden über Transportvesikelstatt (Sekretions- und Endocytoseweg) statt.
Bedingung für den Austritt von Proteinen aus dem ER ist Vollständigkeit und korrekte Faltung. Andernfalls wird das Protein ausgesondert und abgebaut. Dabei spielen Chaperone eine wichtige Rolle, die an unvollständig oder falsch gefaltete Proteine binden und deren Weitertransport verhindern.
Proteine, die nicht weiter transportiert werden, sondern im ER-Lumen verbleiben, werden als ER-ständige Proteine bezeichnet. ER-ständige Proteine sorgen z. B. für die richtige Faltung vieler Proteine, die in das Lumen des ERs gelangen. Ein Beispiel für ein solches ER-ständiges Protein ist die Protein-Disulfid-Isomerase (PDI), die die Bildung von Disulfid-(S-S)-Brücken katalysiert. Ein weiteres ER-ständiges Protein ist das Bindeprotein (BiP), das mit den Hsp70-Proteinen, also Hitzeschockproteinen, verwandt ist und als Chaperon dient. Durch die Bindung an BiP kann die Ausschleusung ER-ständiger Proteine aus dem ER sowie deren Aggregation verhindert werden.
Funktionen des Golgi-Apparates
Der Golgi-Apparat empfängt Proteine und Lipide aus dem ER. Die posttranslationale Proteinmodifizierung, die im ER begonnen hat, wird hier fortgeführt.
Die Übertragung von Oligosacchariden auf bestimmte Asparaginsäurereste der Proteine sowie deren Zurechtschneiden findet schon im ER statt. Nach dieser N-Glykosylierung können sich weitere Modifizierungen im Golgi-Apparat anschließen.
Im Golgi-Apparat findet neben der Glykosylierung von Proteinen auch eine Glykosylierung von Lipiden statt. Dabei entstehen Glykolipide wie Cerebroside und Ganglioside.
Nachdem die Proteinmodifikationen im Golgi-Apparat beendet sind, schließt sich im Trans-Golgi-Netzwerk das Sortieren der Proteine an. Der Transport zu den anderen Kompartimenten des Endomembransystems erfolgt durch kontinuierliche Abschnürung und Fusion von Transportvesikeln. Diese Vesikel verbinden die Kompartimente miteinander, indem sie sich vom Donorkompartiment abschnüren, ihren Inhalt in Richtung Akzeptorkompartiment transportieren, dort mit der Membran verschmelzen und ihren Inhalt freisetzen.
Bei der Vesikelknospung bildet sich eine Hülle aus Proteinen um den knospenden Membranbereich. Es gibt drei verschiedene, gut charakterisierte Hüllentypen, COPI, COPII und Clathrin, sowie zahlreiche andere, über die noch sehr wenig bekannt ist. Vesikel, die COPI und COPII als Hülle tragen, sind am Transport zwischen ER und Golgi-Apparat sowie innerhalb des Golgi-Apparats beteiligt. Dabei transportieren COPII-Vesikel Material vom ER zum Golgi, während COPI-Vesikel den Transport innerhalb des Golgi-Apparates und vom Golgi zum ER erledigen (Abb. 4).
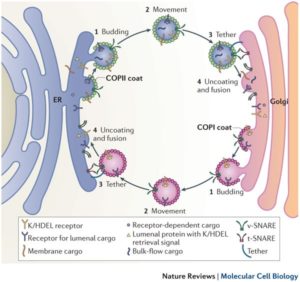
Abb. 4: Transport im Golgi-Apparat
Der Knospungsvorgang wird an Vesikeln durch die Zusammenlagerung der COP-Hüllproteine und Arf, einem kleinen G-Protein, initiiert. Im Cytoplasma liegendiese Komponenten inaktiv vor. Arf trägt gebundenes GDP und auf ein Signal hin wird Arf durch Austausch von GDP gegen GTP aktiviert und lagert sich an der Donormembran an. An dieser Stelle lagern sich die Hüllproteine zusammen und durch die Bildung einer dichten Proteinhülle wird die Knospung vorantreiben bis ein COP-umhüllter Transportvesikel entsteht. Gelangt das Vesikel an die Zielmembran, muss zunächst die Hülle entfernt werden. Das Signal dafür ist die GTP-Hydrolyse an Arf, worauf die Desintegration der gesamten Proteinhülle folgt.
Wie erkennen Vesikel ihr Ziel? Die Spezifität des gerichteten vesikulären Transportes, Proteintargeting, setzt voraus, dass alle Arten von Transportvesikeln in der Zelle auf ihrer Oberfläche Marker besitzen, die Auskunft über ihre Beladung und ihren Ursprung geben. Diese Marker wiederum müssen von komplementären Rezeptoren auf der entsprechenden Zielmembran erkannt werden. An diesem Erkennungsmechanismus ist eine Familie von Membranproteinen beteiligt, die SNAREs (soluble NSF attachment proteins).
In den Vesikeln gibt es die v-SNARES und auf der Zielmembran die t-SNARES. Treffen beide aufeinander (es bildet sich ein trans-SNARE-Komplex), kommt es zu einer Konformationsänderung und nähert die beiden Membranen so sehr aneinander an, dass es zur Fusion kommt. Der gerichtete vesikuläre Transportwird zusätzlich durch rab-Proteine kontrolliert. Sie prüfen, ob jedes Vesikel mit der richtigen Zielmembran fusioniert. Diese kleinen G-Proteine sind nicht nur am vesikulären Transport, sondern auch an Endo- und Exocytosevorgängen beteiligt.
Nach erfolgter Fusion von Vesikel und Zielmembran befindet sich der trans-SNARE-Komplex vollständig in der Zielmembran. Hier interagiert er mit einem Fusionsprotein namens NSF (N-Ethylmaleimid-sensitiven Fusionsprotein) und weiteren Fusionsproteinen. Die Interaktion von NSF mit dem trans-SNARE-Komplex führt unter ATP-Verbrauch zur Dissoziation der trans-SNARE-Komplexe, sodass die v-SNAREs wieder frei vorliegen. Sie werden aus der Ziel-membran zurückgeführt und stehen dann für neue Fusionsrunden zur Verfügung (Abb. 5).
![]()
Abb. 5: Vesikelbildung im GA
Rolle des Cytoskeletts beim Transport
Das Cytoskelett spielt beim Transport von Vesikeln eine wichtige Rolle, allen voran die Mikrotubuli, über deren Aufbau und Rolle wir bei der Mitose einiges erfahren haben.
Sie sind dabei in der Lage, Vesikel durch die Zelle zu transportieren. Die Vesikel enthalten meist verschiedenste Signalstoffe, die zu den unterschiedlichen Zellorganellen transportiert werden.
Die Mikrotubuli legen eine Art „Schienensystem“ für die Vesikel, durch das sie sich bewegen können. Die Vesikel binden sich dabei über sogenannte Motorproteine (Kinesin, Dynein, Myosin) an die Mikrotubuli. Ein solches Motorprotein besteht grundsätzlich aus einer sogenannten Kopfregion mit zwei Köpfen und einer Schwanzregion. Die Kopfregion ist in der Lage, an die Mikrotubuli zu binden und Energie in Form von ATP zu produzieren. Die Schwanzregion bindet die Vesikel an sich. Durch das Spalten von ATP zu ADP wird Energie frei, welche die räumliche Anordnung der Kopfregion verändern kann. Das Motorprotein bewegt sich so schrittweise nach vorne, wobei immer einer der beiden Köpfe an den Mikrotubulus gebunden ist (Abb. 6).

![]()

Abb. 6: Vesikeltransport
Einen wichtigen Vorgang, den man sich merken sollte, ist die sogenannte Exocytose. Bei ihr „verschmelzt“ zuerst das Vesikel mit der Zellmembran und gibt danach ihren Inhalt frei.
Die Exozytose (Abb. 7) dient dazu, Abfall- und Nebenprodukte von Stoffwechselvorgängen aus der Zelle zu schleusen. Außerdem können die Zellen über Exozytose auch nützliche Stoffe nach außen abgeben. Das können zum Beispiel Neurotransmitter oder Hormone sein, die eine reibungslose Kommunikation zwischen mehreren Zellen ermöglichen.
Prinzipiell kann man zwischen einer konstitutiven und einer stimulierten Exozytose unterscheiden. Bei der stimulierten Exozytose ist ein Reiz (z.B. Hormon) für die Ausschüttung der Substanzen verantwortlich, was bei der konstitutiven Exozytose nicht das Fall ist. Sie erfolgt ohne äußere Aktivierung und sorgt zum Beispiel dafür, dass die Biomembranen erweitert werden können.
Die Exozytose steht im Gegensatz zu der Endozytose, die eine Aufnahme von Vesikeln in das Zellinnere beschreibt.
Die Aufnahme von großen Partikeln oder ganzen Zellen nennt man Phagocytose. Sie ist bezeichnend für Zellen mit hoher phagocytotischer Aktivität (z.B. Amöben, viele Ciliaten, Makrophagen, neutrophile Granulocyten).
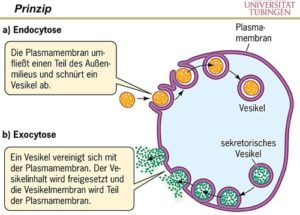
Abb. 7: Edo- und Exozytose
Lysosomen
Die dritte Funktion des Golgi-Apparates ist die Bildung von sogenannten Lysosomen. Diese sind Zellorganellen in eukaryotischen Zellen. Grundsätzlich ähneln sie in ihrem Aufbau den Vesikeln sehr. Sie enthalten verschiedenste Enzyme, die die Verdauung unterstützen können.
In den pflanzlichen Zellen hat der Golgi-Apparat noch eine weitere wichtige Funktion. Hier produziert er verschiedenste Polysaccharide, also Kohlenhydrate. Diese kann die Pflanzenzelle verwenden, um ihre Zellwand aufzubauen.
Ein Lysosom hat eine rundliche Form und ist von einer Membran umgeben.
Der pH-Wert in seinem Inneren (= Lumen) ist sauer und liegt in etwa in einem Bereich um 4,5 – 5. Das Cytosol, in dem sich die Lysosomen bewegen, hat einen neutralen pH-Wert von ca. 7,2 (Abb. 8).
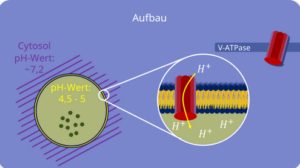
Abb. 8: Lysosom
In diesem sauren Milieu weisen die verschiedenen Enzyme wie zum Beispiel Lipasen, Proteasen und Nukleasen eine hohe Aktivität auf. Diese sogenannten hydrolytischen Enzyme sind in der Lage, Lipide, Proteine, Nukleinsäuren und Polysaccharide durch eine Hydrolyse zu spalten. Ohne die saure Umgebung könnten diese Enzyme nicht effektiv arbeiten, die genannten Stoffe nicht spalten und somit könnte das Lysosom die Stoffe nicht verdauen.
Um den niedrigen pH-Wert zu gewährleisten, benötigt das Lysosom das Transportprotein V-Typ-ATPase, welches in die Lysosommembran eingebettet ist. V-Typ-ATPase ist in der Lage, ATP zu hydrolysieren und dadurch Protonen (H+) vom Cytosol durch die Membran zu „pumpen“. Da der pH-Wert mit steigender Anzahl an H+-Ionen niedriger wird, entsteht so eine durchgängig saure Umgebung im Lysosom (Abb. 9).
Für die Entstehung der Lysosomen ist im Voraus eine Bildung der lysosomalen Enzyme notwendig. Diese Bildung findet im rauen Teil des Endoplasmatischen Retikulums statt. Dort werden sie außerdem in Vesikel gebunden und zum Golgi-Apparat transportiert (Abb. 9).
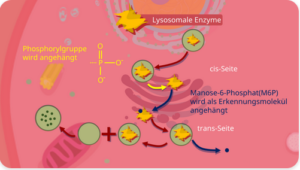
Abb. 9: Lysosom-Entstehung
Auf der cis-Seite des Golgi-Apparates findet eine Vorbereitung für den Weitertransport der lysosomalen Enzyme statt. Dabei wird eine Phosphorylgruppe (PO32-) an das Enzym angehängt (= Phosphorylierung). Danach wird diesen phosphorylierten Enzymen auf der trans-Seite des Golgi-Apparates ein Marker namens Mannose-6-Phosphat (M6P) gegeben. Dieser Marker dient als Erkennungsmolekül für lysosomale Enzyme (Abb. 9).
Im nächsten Schritt werden die lysosomalen Enzyme in Vesikel verpackt. So entstehen die sogenannten primären Lysosomen. Der M6P Marker löst sich ab und kann wiederverwendet werden.
Alle Proteine, die im Golgi-Apparat nicht phosphoryliert wurden, erhalten den M6P-Marker auch nicht. So erkennt die Zelle, dass es sich dabei nicht um lysosomale Proteine handelt. Diese Proteine werden dann in Transportvesikel verpackt und durch Exocytose aus der Zelle herausbefördert.
Die wichtigste Funktion eines Lysosoms besteht darin, zelleigene und zellfremde Stoffe abzubauen. Dafür schließt es den zu verdauenden Stoff (Protein, …) in seinem Innenraum ein. Ein Lysosom ist auch in der Lage, zelleigene Stoffe, wie zum Beispiel Teile von Organellen oder des Cytosols, zu verdauen (= Autophagie). Diese können danach bei Bedarf sogar wiederverwertet werden. Das Lysosom ist also hier eine Art Reparaturvesikel für unsere Zellen.
Peroxisomen
Als letztes seien die Peroxisomen erwähnt (Abb. 10). Ein Peroxisom (eng. peroxisome) ist ein Zellorganell in eukaryotischen Zellen. Peroxisomen verdanken ihren Namen ihrer Fähigkeit, Wasserstoffperoxid abzubauen.
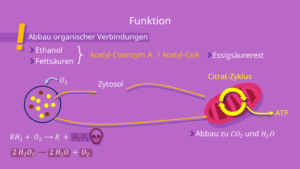
Abb. 10 Peroxisom und seine Funktionen
Eine Funktion des Peroxisoms ist der oxidative Abbau organischer Verbindungen. Das können zum Beispiel Ethanol oder Fettsäuren sein. Insbesondere Ethanol wird vermehrt in den Leberzellen durch Peroxisomen abgebaut.
In tierischen Zellen werden die Fettsäuren und das Ethanol zum sogenannten Acetyl- Coenzym A, kurz CoA, oxidiert. Diesen haben wir bei der Zellatmung kennengelernt.
Jedoch benötigen die Enzyme des Peroxisoms für den Abbau dieser organischen Verbindungen Sauerstoff als zusätzliches Substrat. Durch diesen Sauerstoff kann sich in der Zelle Wasserstoffperoxid (H2O2) als Nebenprodukt bilden.
Wasserstoffperoxid gilt als Zellgift für alle Zellorganellen und insbesondere für das Cytoplasma.
Dieses Zellgift kann das Peroxisom durch seine nächste Funktion auflösen. Die Katalase in seinem Lumen ist in der Lage, Wasserstoffperoxid in die ungefährlichen Moleküle Wasser (H2O) und Sauerstoff (O2) aufzutrennen.
Somit dient das Peroxisom als Reaktionsraum, um die Zelle zu entgiften.
Die dritte Funktion der Peroxisomen bezieht sich auf die Synthese der Myelinscheide von Nervenzellen. Myelin ist eine fetthaltige Biomembran. Wenn diese die Axone der Nervenzellen von Wirbeltieren umwickelt, entsteht die Myelinscheide.